plasmid DNAの物性
2本鎖DNAを輪っか状にデザインした物をPlasmid DNA (pDNA)と言います。完全なpDNAでは、輪ゴムをよじった状態(super coil)となり輪っか状ではなくなります。そのことで、見かけ上の分子量は小さくなります。もしも、2本の内1本に切断箇所(ニック)が存在すると、super coilではなくなり、輪っか状になります。さらに、もう一方の鎖に切断箇所があると、輪っか状も崩れて、直鎖のDNAの形態になります。完全体であるsuper coilのpDNAは、以上の状態の変化による性質の違いを利用して精製されます。
ベクター・デザイン
既存のベクターをデザインするには、既存の知られた遺伝子部品を組み合わせることが基本です。リンクした以下のサイトは、標準のプラスミド・ベクターや、AAVベクター、レンチウイルス・ベクターなど、数十種類のベクターをバックボーンにして、好みのベクターをデザインすることができます。
ベクター・ビルダー
https://www.vectorbuilder.jp/design.html
plasmid DNAの抽出方法
plasmid DNA (pDNA)の製造で最もクリティカルな工程は、その抽出です。pDNAを生産する細胞にE.coliを用いた場合、E.coliのgenomeと目的のpDNAを効率よく分離抽出することが重要です。pDNAは、スーパーコイル(輪ゴムが更によじれている物をイメージするとわかりやすい)となっており、E.coliのgenomeとは物理的強度が異なることを利用して、アルカリ抽出やガラスピーズのミル抽出が一般的に行われます。
- pDNAには耐性があると言って、アルカリ抽出でもガラスビーズ・ミル抽出でも分解されない訳ではないため、pDNAを効率よく抽出するためには、その処理条件の最適化が必要となる
- pDNAは負電荷であるため、その精製は、AEXが基本となるが 12)、疎水クロマト(HIC)も適応できる。
- silica単体にも塩基性アミノ酸バッファで結合させることが可能である
- 工業的な生産では、沈殿化工程は、遠心機を使用するよりフィルター処理する方法が好まれる。フィルター工程の最適化も重要な検討項目である
生産フロー
ベーリンガー社が、CDMOとしてpDNAの効率的な製造方法を考案している。参考文献 3)、その主たる内容は、1~200L規模のcGMPに適用できるpDNAの製造において、高い効率で抽出できるアルカリ抽出およびビーズを用いた方法である。
- Vectorのデザイン
- copy数
- plasmidサイズ (不要な配列の除去)、可能な限り小さく
- 耐性遺伝子から栄養要求遺伝子への変更
- Cell Bankの作成
- 拡大培養
- 生産培養
- ハーベスト
- アルカリ溶解 (Alkaline Lysis)
- pH12
- ガラスビーズ in Tubeによる破砕(マイルドに!)
- supercoiled plasmidは、機械的ストレスに弱い
- 中和(上清にplasmid、沈殿にタンパク質やゲノムDNA)
- pH12
- ろ過システムによる濾過
- 清澄システムにもガラスビースが充填されている
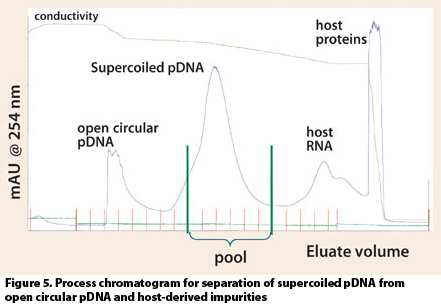
- クロマトグラフィー
- HIC
- Capturing step for pDNA
- binding and elution condition
- removing of endotoxin RNA, genome DNA

- AEX
- binding for pDNA
- removing of entoxine
- SEC
- recovery : 1mg/mL
- HIC
- UF/DF
- concentration : 10mg/mL, but higher viscocity
- Bulk Fill
plasmidの培養方法
参考文献 1)より。大腸菌を使って生産させるのが一般的です。工業的には生産性が高いためです。
| Item | pVGXI1, pVGXI2 |
| Size | 4.2kb, 4.6kb |
| bad case | OD < 15, 0.6g plasmid/10L fermentor |
| optimization | OD >40, 2.4 g ~ 3.0 g plasmid/10L fermentor |
| 考慮事項 | pVGXI2は、高いpoly A, 低いGC比率 |
その他の精製方法として、以下のフローも参考の一つとして示します。特記する点は、ろ過助剤により細胞を効率的に回収している点である。
- 培養条件 : 詳細は参考文献1)を参照
- 流下培養、生産性 0.24g/L
- ハーベストには、ろ過膜、ろ過助剤(珪藻土+ミネラル;ベントナイト)を使用。
- 抽出・溶解工程
- pDNAの選択的沈殿化
- 再溶解/ろ過
参考文献 1)
1) Large Scale Production and Scale Up of DNA Plasmid Vectors With Complex Gene Inserts (2014)
プラスミドベクターを大腸菌で大規模に生産するために、(1)細胞株の最適化と (2)配列の最適化を実施した。
プラスミドpVGXI1(4.2kb)は、最初は10L発酵スケールで増殖は不良(OD <15、プラスミド収量:0.6g / 10L)であった。 細胞株の最適化により改善することができたOD> 40、プラスミド収量2.4g / 10L)。 このプロセスは、100Lおよび400Lスケールまで効果的にスケールアップ可能であった。
プラスミド、pVGXI2(4.6kb)は、その配列の高いA残基の繰り返しと低いGC%で構成されていたが、 3つの異なる大腸菌細胞株で10Lスケールで検討した結果、各細胞株では、増殖は不良(OD <10)であり、プラスミドがほとんど生成されなかった(プラスミド収量:0.7g / 10L)。
配列の最適化を行うことにより、pVGXI2は、標準的な大腸菌株で高い増殖性(OD> 50)およびプラスミド収量(3g / 10L)を10Lスケールで確認できた。
さらに、プロセス最適化により、これらの2つのプラスミドは、発酵収率が4倍に改善し、スケールアップも達成できた。さらに、これらのプラスミドの精製により、臨床使用に適した高品質のDNA産物を得ることができた。
https://www.cell.com/molecular-therapy-family/molecular-therapy/fulltext/S1525-0016(16)35244-3
参考文献 2)
2) 医薬品グレードの大規模プラスミドDNA製造プロセス(2015)
プラスミドDNAの製薬用途には、直接的または間接的に、特定の品質基準が必要です。ヒトの「適正製造基準」(GMP)グレードへの直接遺伝子導入は必須ですが、ウイルスベクター(AAVなど)などのGMP生産では、使用するプラスミドDNAを必ずしもGMPで生産する必要はないとの記載があります。このような規制の側面に加えて、研究室規模(最大数ミリグラム)から工業規模(ミリグラムからグラム規模)へのプラスミドDNA生産プロセスの拡大がここで扱われる問題です。
https://pubmed.ncbi.nlm.nih.gov/24715291/
参考文献 3)
3) プラスミドDNAの工業生産 (2008)
ベーリンガーの新しいcGMP生産システムに関する。pDNAの抽出方法は、アルカリ抽出法とビーズミル抽出法の併用であると理解しました。本当のところ、詳細については不明なのでわかりません。
プラスミドDNAを製造する場合、適正製造基準は慎重なベクター設計から始まります(図1)。真核生物のプロモーター、遺伝子配列、およびポリA部位は主に治療効果に影響を与えますが、ベクターの残りの部分は製造にとって重要です。
ベクターバックボーンのすべての要素、機能、および特性は、プロセスの堅牢性と製品の品質に関して評価する必要があります。
ベーリンガーインゲルハイムオーストリアは、天然のColE1 / pUCoriに基づいた抗生物質を含まないプラスミド選択のためのホストベクターシステムを開発しました。
生産性は、2.4g/L
supercoiled plasmidについての記載あり。培養終了時には、90% supercoiled plasmidであるpDNA均一性を目標とする。
https://www.genengnews.com/magazine/86/industrial-manufacturing-of-plasmid-dna/