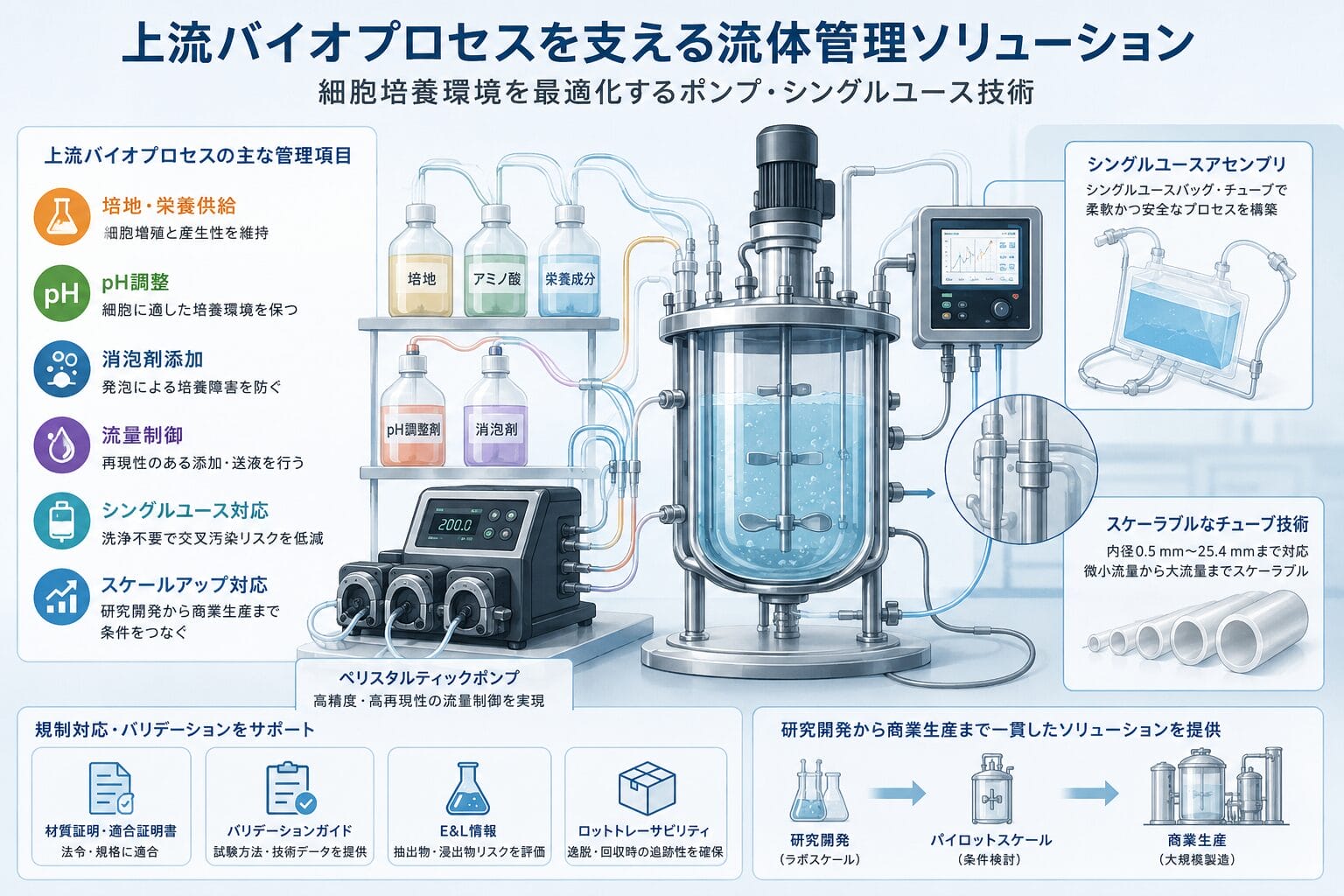
― 品質・生産性・環境負荷を同時に考える時代へ ―
バイオ医薬品の製造では、品質、安全性、安定供給が最優先であることに変わりはありません。一方で、近年は製造工程における水、エネルギー、原材料、廃棄物、施設ユーティリティの使用量をどのように抑えるかも、重要な課題になっています。
バイオプロセスにおけるサステナビリティとは、単に「環境に良い材料を使う」という意味ではありません。開発初期のプロセス設計、設備選定、シングルユース技術の採用、洗浄工程、空調、精製工程、廃棄物処理までを含めて、製品品質を維持しながら、製造全体の環境負荷を下げる考え方です。
Thermo Fisher Scientific の解説では、バイオプロセスのサステナビリティを、製品設計、調達、製造、輸送、使用後のリサイクル・廃棄まで含めて評価する取り組みとして説明しています。特に、プロセス設計の早い段階での判断が、その後の水使用量、エネルギー消費、洗浄薬液、施設面積、HVACなどの負荷に影響するとされています。
なかでも、バイオ医薬品製造における水の問題は避けて通れない重要課題です。細胞培養、培地・バッファー調製、精製、洗浄、滅菌、冷却、蒸気発生など、多くの工程で大量の水が使用されます。また、医薬品製造では単に水の量が確保できればよいわけではなく、精製水やWFIなど、用途に応じた水質を安定的に維持する必要があります。そのため、製造所の立地を考える際には、水資源の量だけでなく、水質、供給安定性、排水処理能力、災害時のリスク、地域社会との水資源共有も含めて検討することが重要です。
したがって、バイオプロセスのサステナビリティでは、水使用量を削減する工程設計、洗浄・滅菌負荷の低減、シングルユース技術の適切な活用、バッファー調製量の最適化などを通じて、品質を維持しながら水資源への依存を下げる取り組みが求められます。
1. なぜバイオプロセスでサステナビリティが重要なのか
バイオ医薬品の製造では、細胞培養、培地調製、バイオリアクター運転、ハーベスト、クロマトグラフィー、UF/DF、充填、洗浄、滅菌、保管など、多くの工程で大量の水とエネルギーが使用されます。
特に大きな負荷となりやすいのは、以下のような項目です。
| 項目 | 環境負荷につながる主な要因 |
|---|---|
| 水 | WFI、精製水、CIP、SIP、洗浄、バッファー調製 |
| エネルギー | HVAC、冷却、加温、滅菌、圧縮空気、冷蔵・冷凍保管 |
| 原材料 | 培地、バッファー、樹脂、フィルター、シングルユースバッグ |
| 廃棄物 | プラスチック廃棄物、フィルター、培養廃液、包装材 |
| 施設 | クリーンルーム面積、空調負荷、ユーティリティ設備 |
ここで重要なのは、環境負荷の多くが「製造開始後」ではなく、プロセス設計と設備選定の段階でほぼ決まってしまう点です。たとえば、ステンレス固定設備を中心にするのか、シングルユース設備を活用するのか、培養スケールをどうするのか、収率をどこまで高めるのか、といった判断が、長期的な水・エネルギー・廃棄物量に影響します。
ISPEも、医薬品製造におけるサステナビリティを、環境影響と健康・安全リスクを評価・管理するためのシステムやプロセスの実装として位置づけています。
2. プロセス設計が資源使用量を左右する
バイオプロセスの環境負荷を下げるうえで、最初に考えるべきなのは「どの材料を使うか」だけではなく、どのようなワークフローにするかです。
たとえば、以下のような設計は資源使用量の削減につながります。
| 設計上の工夫 | 期待される効果 |
|---|---|
| 高密度培養 | 同じ設備面積でより多くの産物を得られる |
| 収率改善 | 1 gあたりの水・エネルギー・原材料使用量を低減 |
| 工程時間短縮 | HVAC、撹拌、冷却、保管などの稼働時間を低減 |
| 閉鎖系プロセス | 清浄度管理、汚染リスク、作業負荷の低減 |
| シングルユース化 | CIP/SIP、水、蒸気、洗浄薬液の削減 |
| 工程統合 | 中間保管、移送、洗浄、設備占有時間の削減 |
Thermo Fisher のページでも、細胞密度の向上、収率改善、バッチサイクル短縮により、施設の生産性を高めつつ、バッチあたりの資源使用量を削減できると説明されています。また、上流工程の改善が、ハーベスト、精製、充填工程の負荷低減にもつながる点が強調されています。
3. シングルユース技術は本当に環境に良いのか
バイオプロセスのサステナビリティでよく議論されるのが、シングルユース技術です。
シングルユースバッグ、チューブ、フィルター、ミキサー、バイオリアクターなどは、使用後に廃棄物が発生します。そのため、一見すると環境負荷が高いように見えます。
しかし、固定式ステンレス設備では、洗浄、滅菌、CIP、SIP、WFI、蒸気、洗浄バリデーション、設備占有時間が必要になります。これに対してシングルユース技術では、洗浄・滅菌に関わる水やエネルギーを削減でき、施設面積やユーティリティ設備を小さくできる場合があります。
つまり、シングルユース技術の評価では、単に「プラスチック廃棄物が出るかどうか」だけでなく、ライフサイクル全体での評価が必要です。LCA、すなわちライフサイクルアセスメントは、原材料調達、製造、輸送、使用、廃棄までを含めて環境影響を評価する手法です。ISPEは、LCAがサービス、設備、工程、製品の排出量、エネルギー、活動量を特定する科学的手法として主流になってきていると説明しています。
一方で、シングルユース技術の弱点は使用後の処理です。多くの部材は複合材料であり、バイオハザード性の有無、汚染状態、材質構成、施設の分別能力、地域の廃棄物処理インフラによって、リサイクルの可否が変わります。BPSAの資料でも、培地・バッファー調製や保管に使われるバッグは比較的リサイクル検討の対象になりやすい一方、使用後バッグの危険性、分別、機械的リサイクル性、再生材の品質などが課題として挙げられています。
4. 「バイオベース材料」と「Greener by Design」
リンク先では、サステナビリティを高めるアプローチとして、Biobased solutions と Greener by Design が示されています。
バイオベース材料とは、石油由来原料の一部または全部を、植物由来などの再生可能資源に置き換えた材料を指します。バイオプロセスで使用されるプラスチック製品や消耗品に、こうした材料を取り入れることで、化石資源への依存を下げられる可能性があります。
ただし、バイオベース材料であれば常に環境負荷が低いとは限りません。原料栽培、土地利用、加工エネルギー、輸送、廃棄方法まで含めて評価する必要があります。したがって、実務上は「バイオベース」という表示だけで判断せず、LCA、供給安定性、抽出物・浸出物、GMP適合性、変更管理への影響を確認することが重要です。
Greener by Design は、製品やプロセスを設計する段階から環境負荷を低減する考え方です。バイオ医薬品製造に置き換えると、以下のような考え方になります。
| 観点 | 実務上の例 |
|---|---|
| 材料設計 | 必要以上に厚いバッグや過剰包装を避ける |
| 工程設計 | 洗浄回数、移送回数、中間保管を減らす |
| 設備設計 | 小型・閉鎖系・モジュール型設備を活用する |
| 品質設計 | 不要な過剰規格や過剰試験を避ける |
| 廃棄設計 | 分別しやすい材料構成にする |
5. プロセス最適化は環境負荷低減そのもの
バイオプロセスでは、収率向上や工程時間短縮は、通常はコスト削減や生産性向上の観点で語られます。しかし、同時に環境負荷低減にも直結します。
たとえば、同じ設備、同じ培地量、同じ空調条件で、抗体の収量が2倍になれば、製品1 gあたりの水、電力、培地、バッファー、フィルター、樹脂、廃棄物の負荷は相対的に下がります。
この意味で、サステナビリティは環境部門だけの課題ではなく、プロセス開発、製造技術、CMC、QA、QC、エンジニアリング、購買、サプライチェーンが関与するテーマです。
特に重要なのは、以下のようなKPIです。
| KPI | 意味 |
|---|---|
| 収率 | 投入資源に対する目的物の回収効率 |
| 培養密度 | 設備容量あたりの生産性 |
| バッチサイクルタイム | 設備・空調・作業時間の使用効率 |
| 水使用量 | WFI、精製水、洗浄水を含む |
| エネルギー使用量 | HVAC、冷却、加温、滅菌など |
| 廃棄物量 | プラスチック、フィルター、培地・バッファー廃液 |
| PMI | Process Mass Intensity。製品量あたりの投入物量 |
BioPhorumも、バイオ製造における環境サステナビリティでは、バリューチェーン全体での協働が必要であり、各部門が自分たちの工程で改善を進める必要があるとしています。
6. GMPとの関係:環境負荷低減だけを優先してはいけない
バイオ医薬品製造では、サステナビリティの取り組みは重要ですが、GMP上の品質保証を犠牲にすることはできません。
たとえば、以下のような変更は、単なる環境対策ではなく、GMP上の変更管理対象になります。
| 変更例 | GMP上の確認事項 |
|---|---|
| シングルユースバッグの材質変更 | 抽出物・浸出物、適合性、供給者管理 |
| フィルター変更 | 完全性試験、保持性能、バリデーション影響 |
| 洗浄条件の短縮 | 残留物、洗浄バリデーション、交叉汚染リスク |
| バッファー濃縮化 | 溶解性、安定性、混合均一性 |
| 工程時間短縮 | 不純物、HCP、DNA、凝集体、微生物リスク |
| 廃棄方法変更 | バイオハザード、法規制、施設ルール |
サステナビリティ施策は、QA、製造、QC、エンジニアリング、EHS、購買、サプライヤー品質管理を含めて評価する必要があります。特に商用製造では、承認事項、バリデーション、規格、供給者変更、年次照査への影響も確認すべきです。
7. 実務で使えるチェックリスト
バイオプロセスのサステナビリティを検討する際には、以下のような観点で整理すると実務に落とし込みやすくなります。
開発初期
| 確認項目 | ポイント |
|---|---|
| 工程数を減らせるか | 不要な中間工程・保管を削減 |
| 収率を改善できるか | 製品1 gあたりの資源使用量を低減 |
| 閉鎖系にできるか | 汚染リスクと清浄度管理負荷を低減 |
| シングルユース化できるか | 洗浄・滅菌負荷を低減 |
| 材料の標準化が可能か | 在庫、廃棄、供給リスクを低減 |
製造設計
| 確認項目 | ポイント |
|---|---|
| 水使用量を見積もっているか | WFI、精製水、CIP、SIPを含める |
| HVAC負荷を評価しているか | クリーンルーム面積とグレードが影響 |
| バッファー量を削減できるか | 濃縮、インライン希釈、工程統合 |
| 廃棄物の分別が可能か | リサイクル可能性を事前に確認 |
| サプライヤーの環境情報があるか | 材料由来、LCA、リサイクル対応 |
商用製造
| 確認項目 | ポイント |
|---|---|
| 変更管理に入っているか | 品質影響評価が必要 |
| バリデーション影響はあるか | 洗浄、工程、分析法、設備に注意 |
| 当局申請への影響はあるか | 承認事項との関係を確認 |
| KPIを継続監視しているか | 水、エネルギー、廃棄物、収率 |
| 年次照査に反映しているか | 継続的改善として管理 |
8. 今後の方向性
今後のバイオ医薬品製造では、サステナビリティは「企業イメージ」だけの話ではなく、製造コスト、供給安定性、設備投資、規制対応、社会的責任に関わる実務課題になります。
特に重要になるのは、以下の3点です。
第一に、プロセス開発段階から環境負荷を評価することです。後から廃棄物処理だけを改善するよりも、工程そのものを効率化する方が効果的です。
第二に、シングルユース技術を単純に善悪で判断しないことです。廃棄物は課題ですが、水、エネルギー、洗浄、施設面積を含めたライフサイクル全体で評価する必要があります。
第三に、GMPとサステナビリティを両立させることです。品質保証、バリデーション、供給者管理、変更管理を踏まえたうえで、現実的な改善策を積み上げる必要があります。
バイオプロセスのサステナビリティとは、環境負荷を下げるだけでなく、より効率的で、堅牢で、将来にわたって継続可能な製造体制を作る取り組みだといえます。