Post Views: 4
はじめに
iPS細胞由来製品、体性幹細胞加工製品、免疫細胞加工製品、遺伝子治療用製品などは、薬機法上の「再生医療等製品」として扱われます。
再生医療等製品では、製造所における製造管理・品質管理について、医薬品GMP省令ではなく、GCTP省令 が適用されます。
ここで誤解しやすいのが、GMPとGCTPの関係です。
再生医療等製品では、制度上、「GMPにGCTPを加味する」というよりも、再生医療等製品の製造管理・品質管理基準としてGCTP省令に基づく体制を構築する と表現する方が正確です。
ただし、GCTPはGMPと無関係ではありません。GCTPには、製造管理、品質管理、文書・記録管理、逸脱管理、変更管理、教育訓練、自己点検、バリデーション、品質リスクマネジメントなど、GMPと共通する品質システムの考え方が多く含まれています。
そのため、実務上は、GMP的な品質保証の考え方を踏まえながら、再生医療等製品の特性を反映したGCTP体制を整備する と理解するとよいでしょう。
さらに重要なのは、GCTPが適用されるからといって、製造販売業者のGQP体制が不要になるわけではないという点です。
結論として、再生医療等製品でも、製造販売業者にはGQP省令に基づく品質管理体制が必要です。
この記事の結論
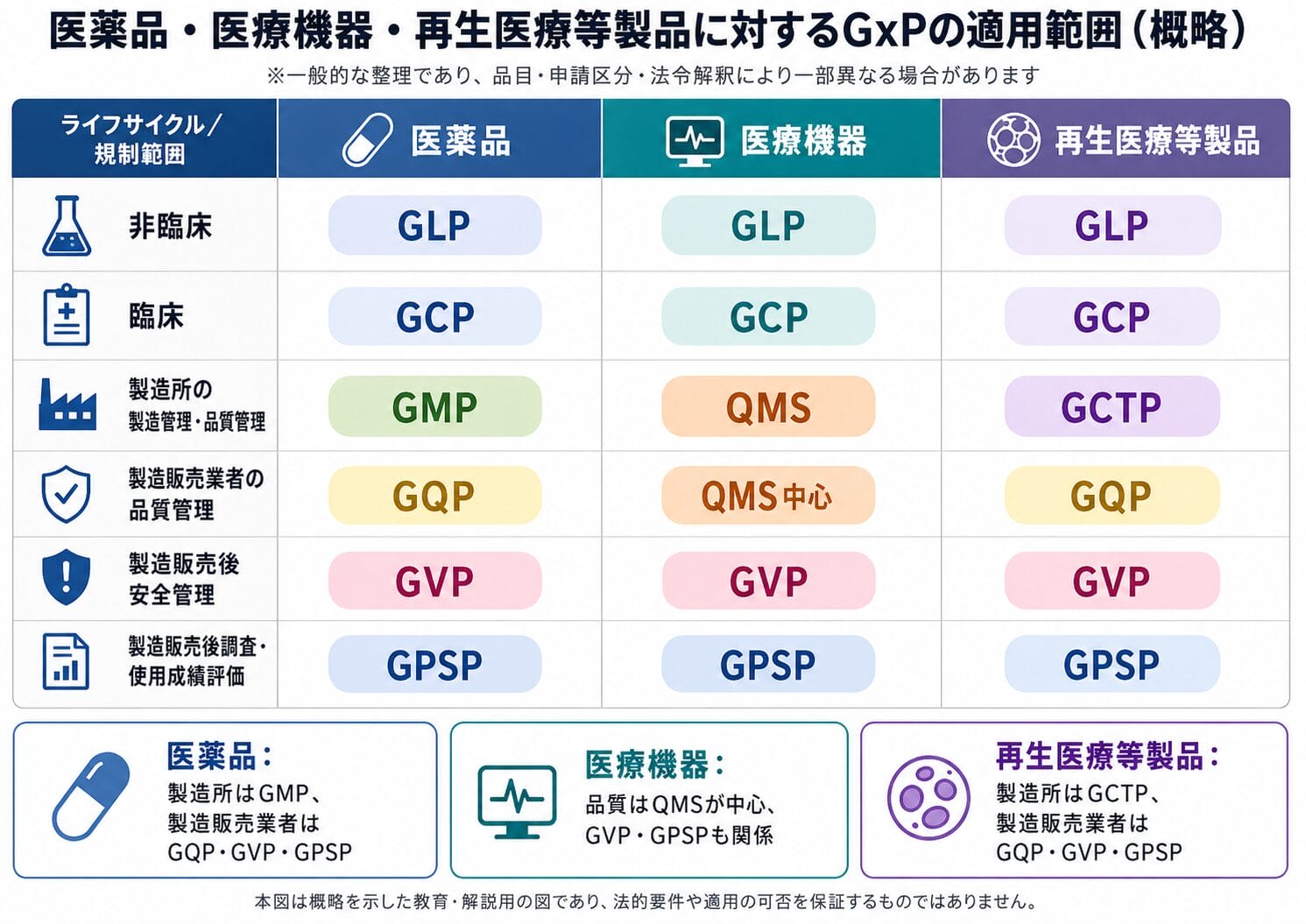
再生医療等製品の品質・安全性管理は、次のように整理すると理解しやすくなります。
領域 主な対象 適用される基準 医薬品・医薬部外品の製造所における製造管理・品質管理 医薬品等の製造業者、製造所 GMP省令 再生医療等製品の製造所における製造管理・品質管理 再生医療等製品の製造業者、外国製造業者、製造所 GCTP省令 製造販売業者の品質管理・品質保証 製造販売業者 GQP省令 製造販売後安全管理 製造販売業者 GVP省令 製造販売後調査・使用成績調査等 製造販売業者 再生医療等製品GPSP省令
GMPとGCTPは、どちらも製造所側の製造管理・品質管理基準ですが、対象となる製品区分が異なります。
医薬品ではGMP省令、再生医療等製品ではGCTP省令が対応する基準になります。
一方、GQPは製造販売業者の品質管理基準、GVPは製造販売後安全管理、GPSPは製造販売後調査・使用成績評価に関係する基準です。
つまり、GCTPは製造所側の基準、GQPは製造販売業者側の基準 であり、両者は代替関係ではありません。
再生医療等製品とは何か
再生医療等製品は、薬機法上、医薬品や医療機器とは別に位置づけられた製品区分です。
代表的には、以下のようなものが含まれます。
分類 例 細胞加工製品 iPS細胞由来製品、体性幹細胞加工製品、免疫細胞加工製品など 組織加工製品 培養表皮、培養軟骨など 遺伝子治療用製品 ウイルスベクター製品、遺伝子導入細胞製品など
注意が必要なのは、薬機法上の「再生医療等製品」と、再生医療等安全性確保法に基づいて医療機関で実施される「再生医療」は、制度上の位置づけが異なるという点です。
本記事で扱うのは、薬機法上の再生医療等製品を製造販売する場合のGMP・GCTP・GQP・GVP・GPSPの整理 です。
GMPとGCTPの関係
医薬品では、製造所における製造管理・品質管理についてGMP省令が適用されます。
一方、再生医療等製品では、製造所における製造管理・品質管理についてGCTP省令が適用されます。
したがって、再生医療等製品については、制度上、GMP省令を基本にしてGCTPを上乗せする という構造ではありません。
製造所管理の主たる基準としては、再生医療等製品の特性に応じたGCTP省令 が適用されます。
製品区分 製造所の製造管理・品質管理基準 実務上の理解 医薬品・医薬部外品 GMP省令 医薬品等の製造管理・品質管理基準 医療機器・体外診断用医薬品 QMS省令 医療機器等の品質マネジメント基準 再生医療等製品 GCTP省令 再生医療等製品の特性を踏まえた製造管理・品質管理基準
ただし、GCTPはGMPと共通する考え方を多く持っています。
たとえば、次のような考え方は、GMPとGCTPの双方で重要です。
共通する品質システムの考え方 内容 製造管理 手順に従って一貫した製造を行う 品質管理 試験検査、規格適合性、記録確認を行う 文書・記録管理 手順書、製造記録、試験記録を管理する 逸脱管理 手順や規格からの逸脱を評価し、是正する 変更管理 工程、設備、原材料、試験法等の変更影響を評価する 教育訓練 職員に必要な教育訓練を行う 自己点検 体制や運用状況を定期的に確認する バリデーション 工程や方法が期待どおり機能することを確認する 品質リスクマネジメント 品質リスクを評価し、適切に管理する
このため、実務上は、GMPの経験や品質保証の考え方を活かしながら、再生医療等製品に特有の管理項目をGCTP体制として組み込むことになります。
言い換えると、再生医療等製品で必要なのは、**「GMPにGCTPを加味した体制」ではなく、「GMP的な品質システムの考え方を踏まえたGCTP体制」**です。
この違いは細かいようですが、制度理解としては重要です。
「GMPにGCTPを加味する」と表現すると、GMPが主でGCTPが補足であるように読めます。しかし、再生医療等製品の製造所管理では、主たる基準はGCTP省令です。
GCTP省令とは何か
GCTPは、再生医療等製品の製造管理及び品質管理の基準です。
正式名称は、**「再生医療等製品の製造管理及び品質管理の基準に関する省令」**です。
GCTPは単なるガイドラインではありません。再生医療等製品の製造管理・品質管理について、省令として定められた基準です。
GCTP省令では、再生医療等製品の特性を踏まえ、次のような管理が重要になります。
管理項目 内容 製造管理 製造手順、工程管理、逸脱管理、変更管理など 品質管理 試験検査、規格適合性、記録管理など 無菌性管理 無菌操作等区域、清浄度管理、汚染防止など 原材料管理 細胞、組織、培地、サイトカイン、血清、ベクター、容器等の管理 ドナー管理 ドナー情報、適格性、感染症リスク等 バリデーション・ベリフィケーション 工程、試験法、無菌操作、輸送条件等の確認 品質リスクマネジメント 細胞製品特有のばらつき、不均質性、工程変動等への対応 トレーサビリティ 原材料、細胞、ドナー、ロット、患者等との紐づけ
再生医療等製品では、細胞・組織・遺伝子改変技術を扱うため、医薬品GMPだけでは整理しにくい論点があります。
たとえば、ドナー管理、細胞・組織のトレーサビリティ、無菌操作等区域、細胞製品のばらつき、短い使用期限、凍結・解凍条件、ベリフィケーションなどです。
そのため、GCTPは、GMP的な品質システムの考え方を踏まえつつ、再生医療等製品の特性に合わせて整備された製造管理・品質管理基準と理解するとよいでしょう。
GCTP適合性調査とは何か
再生医療等製品では、GCTP省令に基づき、製造所の管理状況が確認されます。
PMDAは、GCTP適合性調査について、国内外の製造所に対し、製造設備に加えて、製造管理や品質管理の手法がGCTPに適合しているかを調査すると説明しています。
つまり、GCTP適合性調査は、製造所が適切に再生医療等製品を製造できる状態にあるかを確認する調査 です。
PMDAの整理では、医薬品等ではGMP、医療機器等ではQMS、再生医療等製品ではGCTPというように、製品区分ごとに製造所の管理基準が対応しています。
したがって、再生医療等製品の製造所については、GMP適合性調査ではなく、GCTP適合性調査の枠組みで製造管理・品質管理の適合性が確認されます。
ここで重要なのは、GCTP適合性調査が製造所側の管理状況を確認するものであり、製造販売業者のGQP責任を代替するものではないという点です。
GQP省令とは何か
GQPは、製造販売業者が製品の品質を確保するための品質管理基準です。
正式名称は、**「医薬品、医薬部外品、化粧品及び再生医療等製品の品質管理の基準に関する省令」**です。
GQP省令には、再生医療等製品の品質管理の基準が含まれています。
したがって、再生医療等製品を製造販売する場合でも、製造販売業者はGQP省令に基づく品質管理体制を整える必要があります。
GQPで重要になるのは、製造販売業者が、製品を市場に出す立場として品質を管理することです。
具体的には、次のような業務が関係します。
GQP上の主な業務 内容 市場出荷管理 製品を市場に出荷してよいかを判断する 製造業者等の管理監督 製造所、試験委託先、保管・輸送委託先等を管理する 品質情報の処理 苦情、品質不良、逸脱情報などを評価する 品質不良対応 原因調査、是正措置、予防措置を行う 回収処理 回収判断、行政報告、回収実施、再発防止を行う GVP部門との連携 品質問題が安全性問題に発展する場合に連携する
GMP・GCTP・GQPの違い
GMP、GCTP、GQPはいずれも品質に関係しますが、対象と責任範囲が異なります。
GMPとGCTPは、いずれも製造所側の製造管理・品質管理基準です。
区分 GMP GCTP GQP 主な対象 医薬品等の製造所 再生医療等製品の製造所 製造販売業者 主な目的 医薬品等を適切に製造・品質管理すること 再生医療等製品を適切に製造・品質管理すること 製造販売業者として市場出荷を含めた品質を保証すること 主な業務 製造管理、品質管理、逸脱管理、変更管理、バリデーション等 製造管理、品質管理、無菌管理、ドナー管理、トレーサビリティ、ベリフィケーション等 市場出荷、製造業者等の管理、品質情報、品質不良、回収等 位置づけ 製造所側の基準 製造所側の基準 製造販売業者側の基準 再生医療等製品での位置づけ 直接の主基準ではない 製造所管理の主基準 製造販売業者に必要
したがって、再生医療等製品では、製造所側ではGCTP、製造販売業者側ではGQPを整備する必要があります。
GMPの考え方はGCTP体制を理解・構築するうえで有用ですが、制度上は、再生医療等製品の製造所管理の主基準はGCTPです。
製造販売業者と製造業者を分けて考えることが重要
再生医療等製品の品質管理を理解するうえで重要なのは、製造販売業者 と製造業者 を分けて考えることです。
製造業者は、製造所においてGCTP省令に基づき、製造管理・品質管理を実施します。
一方、製造販売業者は、製品を市場に出す主体として、GQP省令に基づき、製造所や委託先を管理し、市場出荷、品質情報、品質不良、回収などを管理します。
立場 主な責任 製造業者 製造所でGCTPに基づく製造管理・品質管理を行う 製造販売業者 GQPに基づき、製造所を管理し、市場出荷と市販後品質を管理する
このため、製造所がGCTPに適合していても、製造販売業者は、製造所の記録、試験結果、逸脱、変更、品質情報、輸送条件などを確認し、製造販売業者として品質を保証する必要があります。
市場出荷判定はGQP側の重要業務
GCTPでは、製造所における製造管理・品質管理が中心になります。
しかし、最終的に製品を市場に出荷してよいかを判断するには、製造販売業者としての確認が必要です。
市場出荷判定では、たとえば次のような情報を確認する必要があります。
確認項目 例 製造記録 指図通りに製造されたか 試験結果 規格に適合しているか 逸脱 品質に影響する逸脱がないか 変更 承認内容や品質に影響する変更がないか 保管条件 温度、時間、凍結条件などが守られているか 輸送条件 輸送中の温度逸脱、時間逸脱がないか 使用期限 出荷時点で使用可能期間が適切に残っているか 無菌性・微生物管理 無菌試験、環境モニタリング等の結果に問題がないか
再生医療等製品では、細胞生存率、細胞数、同一性、純度、力価、無菌性、エンドトキシン、マイコプラズマ、凍結・解凍条件などが品質判断に影響することがあります。
そのため、市場出荷判定は、GCTPとGQPをつなぐ非常に重要な実務ポイントです。
品質取決めと委託先管理も重要
再生医療等製品では、製造工程、試験、保管、輸送、原材料供給などを外部に委託する場合があります。
この場合、製造販売業者は、委託先を選ぶだけでは不十分です。
品質取決めを結び、責任範囲を明確にしておく必要があります。
品質取決めでは、少なくとも次のような事項を整理することが重要です。
項目 内容 責任範囲 製造、試験、保管、輸送、記録、報告の責任 変更連絡 工程、設備、原材料、試験法、委託先変更時の連絡 逸脱報告 品質に影響する逸脱が発生した場合の報告 品質情報 苦情、品質不良、異常傾向の共有 回収対応 回収判断、連絡体制、記録保管 監査 製造販売業者による委託先確認 記録保存 製造記録、試験記録、輸送記録等の保存 トレーサビリティ 原材料、細胞、ドナー、ロット、患者との紐づけ
特に再生医療等製品では、原材料、細胞、ベクター、培地、サイトカイン、血清、容器、凍結保存条件、輸送条件などが品質に大きく影響する可能性があります。
したがって、品質取決めと委託先管理は、GQP上の重要な論点です。
GVPとの連携も必要
再生医療等製品では、品質管理だけでなく、製造販売後安全管理も重要です。
GVPは、製造販売後安全管理の基準です。
再生医療等製品では、有害事象、安全性情報、感染症リスク、免疫反応、腫瘍形成リスク、長期フォローアップなどが重要になる場合があります。
品質問題が安全性問題に発展することもあります。たとえば、無菌性、細胞生存率、力価、異物、輸送温度逸脱などの品質問題が、患者への安全性に影響する可能性があります。
そのため、GQP部門とGVP部門は、別の機能として整理しながらも、実務上は密接に連携する必要があります。
情報の種類 主な管理部門 連携が必要な理由 品質苦情 GQP 安全性に影響する可能性がある 有害事象 GVP 品質不良が原因の可能性がある 回収判断 GQP/GVP 品質・安全性の両面から判断が必要 添付文書改訂 GVP中心 品質情報が安全対策に影響する場合がある 製造工程変更 GQP中心 安全性・有効性への影響評価が必要
GPSP・使用成績評価も関係する
再生医療等製品では、製造販売後調査や使用成績評価も重要です。
特に、条件及び期限付承認制度を利用する再生医療等製品では、承認後に有効性・安全性に関するデータを収集し、通常承認や再審査に向けて評価することが必要になります。
製造販売後には、使用成績調査、製造販売後データベース調査、製造販売後臨床試験などが関係する場合があります。
したがって、再生医療等製品では、承認取得時点だけでなく、承認後のデータ収集、品質情報、安全性情報、使用成績評価まで含めたライフサイクル管理が重要になります。
条件及び期限付承認後に注意すべきこと
再生医療等製品では、条件及び期限付承認が認められる場合があります。
この制度では、承認時点で一定の有効性が推定され、安全性が確認されていることを前提に、承認後に追加データを収集して評価を行うことになります。
そのため、製造販売業者は、承認後も次のような点を管理する必要があります。
項目 内容 有効性データ 実臨床で期待される効果が得られているか 安全性データ 重篤な有害事象、遅発性リスク、長期安全性 品質の持続性 承認時と同等の品質が維持されているか 製造工程変更 変更が品質・有効性・安全性に影響しないか 使用成績評価 承認条件に基づく調査・評価 再審査・通常承認 次の承認段階に向けた資料整備
特に細胞加工製品では、製品の不均質性、ドナー差、製造工程のばらつき、スケール変更、原材料変更などが品質に影響しやすいため、変更管理と同等性・同質性評価が重要です。
実務上の体制イメージ
再生医療等製品の製造販売業者では、少なくとも次のような体制整理が必要になります。
役割 主な責任 総括製造販売責任者 品質管理業務・安全管理業務を含む製造販売業者全体の統括 品質保証責任者 / 品質保証部門 GQP業務、市場出荷、製造業者等の管理、品質情報、回収対応 安全管理責任者 / 安全管理部門 GVP業務、安全性情報の収集・評価、安全確保措置 製造所の製造管理者・品質部門 GCTPに基づく製造所内の製造管理・品質管理 製造販売後調査担当 GPSPに基づく調査、使用成績評価、承認条件評価 薬事担当 承認申請、一変申請、軽微変更届、PMDA相談等 委託先管理担当 製造委託、試験委託、保管・輸送委託の管理
重要なのは、製造所の品質部門と、製造販売業者の品質保証部門を同一視しないこと です。
製造所はGCTPに基づいて製造管理・品質管理を行います。一方、製造販売業者はGQPに基づいて、製造所を管理監督し、出荷可否を判断し、市場で発生した品質問題にも対応します。
まとめ
iPS細胞製品などの再生医療等製品では、製造所における製造管理・品質管理にはGCTP省令が適用されます。
医薬品ではGMP省令が製造所管理の基準になりますが、再生医療等製品ではGCTP省令が対応する基準になります。
したがって、制度上は「GMPにGCTPを加味する」というよりも、再生医療等製品ではGCTP省令に基づく製造管理・品質管理体制を構築する ことが正確です。
ただし、GCTPにはGMPと共通する品質システムの考え方が多く含まれています。そのため、実務上は、GMP的な品質保証の考え方を踏まえながら、再生医療等製品の特性を反映したGCTP体制を整備する と理解するとよいでしょう。
一方、GCTP省令は製造所側の基準であり、製造販売業者の品質管理責任を代替するものではありません。
再生医療等製品であっても、製造販売業者にはGQP省令に基づく品質管理体制が必要です。
さらに、製造販売後安全管理としてGVP、製造販売後調査・使用成績評価としてGPSPも関係します。
つまり、再生医療等製品の品質・安全性管理は、次のように整理できます。
GMPは医薬品等の製造所管理、GCTPは再生医療等製品の製造所管理、GQPは製造販売業者の品質管理、GVPは製造販売後安全管理、GPSPは製造販売後調査・使用成績評価の基準である。
再生医療等製品では、細胞・組織・遺伝子改変技術などを扱うため、原材料管理、ドナー管理、無菌性、保存・輸送、トレーサビリティ、変更管理、製造販売後のデータ収集が特に重要になります。
注意点・例外
この記事は、薬機法上の再生医療等製品に関する制度整理です。
医療機関で実施される再生医療、自由診療、臨床研究、特定細胞加工物の提供などは、再生医療等安全性確保法の対象となる場合があり、薬機法上の再生医療等製品とは制度が異なります。
また、再生医療等製品本体の製造管理・品質管理はGCTP省令で整理されますが、原材料、培地成分、添加剤、ウイルスベクター、併用される医薬品・医療機器、試験用試薬、保管・輸送資材などについては、GMP、QMS、原薬GMP、医薬品添加剤GMP、GDP的管理などが関係する可能性があります。
そのため、個別品目では、再生医療等製品本体はGCTPで管理しつつ、関連する原材料・供給品・併用製品について、どの基準・品質取決め・供給者管理を適用するかを整理する必要があります。
実際のGQP手順書、品質取決め、GVP/GPSP体制、条件及び期限付承認後の調査計画、PMDA相談資料、承認申請資料の作成については、薬事・品質保証・安全管理の専門家に確認が必要です。
参考文献・出典
【確実性: 高】
用語集
用語 読み・英語 意味 本記事での位置づけ 再生医療等製品 さいせいいりょうとうせいひん / Regenerative Medical Products 細胞加工製品、組織加工製品、遺伝子治療用製品など、薬機法上の製品区分 本記事の主対象 iPS細胞由来製品 induced Pluripotent Stem Cell-derived Product iPS細胞から分化誘導した細胞などを用いる再生医療等製品 再生医療等製品の代表例 細胞加工製品 Cell-processed Product 細胞を採取、培養、加工、分化誘導などして製品化したもの GCTP管理の重要対象 遺伝子治療用製品 Gene Therapy Product 遺伝子導入、ウイルスベクター、遺伝子改変細胞などを用いる製品 再生医療等製品に含まれる場合がある 薬機法 医薬品医療機器等法 / PMD Act 医薬品、医療機器、再生医療等製品などを規制する日本の法律 再生医療等製品の制度上の根拠 再生医療等安全性確保法 Act on the Safety of Regenerative Medicine 医療機関で行われる再生医療等の提供計画、細胞培養加工施設などを規制する法律 薬機法上の再生医療等製品とは別制度 GMP Good Manufacturing Practice 医薬品・医薬部外品の製造所における製造管理・品質管理の基準 医薬品側の製造所管理基準 GMP省令 医薬品及び医薬部外品の製造管理及び品質管理の基準に関する省令 医薬品・医薬部外品のGMPを定めた省令 再生医療等製品では主基準ではない GCTP Good Gene, Cellular, and Tissue-based Products Manufacturing Practice 再生医療等製品の製造所における製造管理・品質管理の基準 再生医療等製品の製造所管理の主基準 GCTP省令 再生医療等製品の製造管理及び品質管理の基準に関する省令 再生医療等製品の製造管理・品質管理を定めた省令 本記事の中心用語 GQP Good Quality Practice 製造販売業者が製品品質を確保するための品質管理基準 製造販売業者側の品質管理基準 GQP省令 医薬品、医薬部外品、化粧品及び再生医療等製品の品質管理の基準に関する省令 製造販売業者の品質管理基準を定めた省令 再生医療等製品にも適用される GVP Good Vigilance Practice 製造販売後安全管理の基準 市販後安全性情報、有害事象、安全確保措置に関係 GVP省令 製造販売後安全管理の基準に関する省令 製造販売後の安全管理体制を定めた省令 GQPと連携が必要 GPSP Good Post-marketing Study Practice 製造販売後調査・試験の実施基準 使用成績調査、承認条件評価などに関係 QMS Quality Management System 医療機器・体外診断用医薬品の品質マネジメントシステム GMP/GCTPとの比較対象 QMS省令 医療機器及び体外診断用医薬品の製造管理及び品質管理の基準に関する省令 医療機器等の品質管理基準を定めた省令 再生医療等製品ではなく医療機器側の基準 製造販売業者 MAH / Marketing Authorization Holder 製品を市場に出す責任を負う業者 GQP、GVP、GPSPの主体 製造業者 Manufacturer 製造所で製品を製造する業者 GCTPの主体 外国製造業者 Foreign Manufacturer 海外で製造を行う製造業者 GCTP適合性調査の対象になり得る 製造所 Manufacturing Site 実際に製造、試験、保管などを行う場所 GCTP適合性調査の対象 総括製造販売責任者 Marketing Supervisor-General 製造販売業者における品質管理・安全管理を統括する責任者 GQP/GVP体制の統括者 品質保証責任者 Quality Assurance Manager GQP業務を実務上管理する責任者 市場出荷、品質情報、回収等を管理 安全管理責任者 Safety Management Supervisor GVP業務を管理する責任者 安全性情報、安全確保措置を管理 製造管理者 Manufacturing Manager 製造所における製造管理・品質管理の責任者 GCTP体制の現場責任者 市場出荷判定 Market Release Decision 製品を市場へ出荷してよいかを確認・判断すること GQP側の重要業務 品質取決め Quality Agreement 製造販売業者と製造業者・委託先との責任範囲を定める文書 委託先管理の基本文書 委託先管理 Supplier / Contractor Management 製造、試験、保管、輸送などの委託先を管理すること GQP上の重要業務 GCTP適合性調査 GCTP Compliance Inspection 製造所がGCTPに適合しているか確認する調査 製造所側の確認でありGQPを代替しない GMP適合性調査 GMP Compliance Inspection 医薬品等の製造所がGMPに適合しているか確認する調査 医薬品側の製造所確認 逸脱管理 Deviation Management 手順、規格、承認事項などから外れた事象を評価・処理すること GMP/GCTP/GQPで重要 変更管理 Change Control 工程、設備、原材料、試験法などの変更影響を評価・管理すること 再生医療等製品では特に重要 是正措置・予防措置 CAPA / Corrective and Preventive Action 不具合や逸脱の原因を是正し、再発を防止する活動 品質不良対応の基本 バリデーション Validation 工程や方法が期待どおり機能することを文書化して確認すること GMP/GCTP共通の品質保証要素 ベリフィケーション Verification あらかじめ定めた要求事項を満たしていることを確認すること GCTPで重要な考え方 品質リスクマネジメント Quality Risk Management 品質に関するリスクを評価・低減・管理する考え方 GMP/GCTP共通の重要要素 無菌操作等区域 Aseptic Processing Area 無菌的な作業を行う区域 再生医療等製品で重要 清浄度管理 Cleanliness Control 作業区域の微粒子・微生物汚染を管理すること 無菌性確保に関係 無菌性 Sterility 生菌による汚染がないこと 再生医療等製品の重要品質特性 エンドトキシン Endotoxin グラム陰性菌由来の発熱性物質 投与製品の安全性に関係 マイコプラズマ Mycoplasma 細胞培養で問題となる微生物汚染の一種 細胞加工製品で重要な試験対象 ドナー Donor 細胞や組織を提供する人または動物 原材料・感染症リスク管理に関係 ドナー管理 Donor Control ドナーの適格性、感染症リスク、由来情報などを管理すること GCTP特有の重要論点 トレーサビリティ Traceability 原材料、細胞、ドナー、ロット、患者などの履歴を追跡できること 再生医療等製品で重要 細胞生存率 Cell Viability 製品中の細胞のうち生存している細胞の割合 細胞製品の品質判断に関係 細胞数 Cell Number 製品中に含まれる細胞の数 投与量・規格に関係 同一性 Identity 製品が意図した細胞・成分であること 品質試験の重要項目 純度 Purity 目的外細胞や不純物が少ないこと 品質特性の一つ 力価 Potency 製品が意図した生物学的機能を有すること 有効性に関係する重要品質特性 凍結・解凍条件 Freezing and Thawing Conditions 細胞製品の保存・使用時の温度や手順条件 品質維持に重要 使用期限 Expiry Date / Shelf Life 製品を使用できる期限 細胞製品では短い場合がある 品質情報 Quality Information 苦情、品質不良、逸脱、異常傾向などの品質関連情報 GQPで処理する情報 品質不良 Quality Defect 製品品質に問題がある状態 回収や安全性評価につながる可能性 回収 Recall 市場に出た製品を回収すること GQP/GVP連携が必要 安全性情報 Safety Information 有害事象、副作用、感染症リスクなどの情報 GVPで管理 有害事象 Adverse Event 製品使用後に発生した望ましくない医学的事象 GVPの主要管理対象 安全確保措置 Safety Measures 使用上の注意改訂、情報提供、出荷停止、回収等の措置 GVP上の対応 条件及び期限付承認 Conditional and Time-limited Approval 一定条件のもとで期限を付して承認する制度 再生医療等製品で重要 使用成績調査 Post-marketing Use-results Survey 市販後に実使用下で有効性・安全性を調査すること GPSPに関係 製造販売後データベース調査 Post-marketing Database Study 医療情報データベース等を用いた製造販売後調査 GPSP関連 製造販売後臨床試験 Post-marketing Clinical Trial 承認後に実施する臨床試験 承認条件評価等に関係 使用成績評価 Use-results Evaluation 承認後に収集した実使用データを評価すること 条件及び期限付承認後に重要 同等性・同質性 Comparability 製造変更前後で品質・有効性・安全性が同等または同質と評価できること 工程変更時に重要 ライフサイクル管理 Lifecycle Management 開発、承認、製造、市販後まで一貫して製品を管理する考え方 再生医療等製品で重要
【注意点・例外】
この用語集は、ブログ読者向けにわかりやすく要約したものです。法令上の厳密な定義、通知上の用語、承認申請資料や手順書での記載では、正確な条文・通知・PMDA資料に基づく確認が必要です。
参考文献・出典
再生医療等製品に係るGxPは、日本・米国・EUでかなり整理の仕方が異なります 。日本は「GCTP」という再生医療等製品専用の製造管理・品質管理省令を持つ のに対し、米国はHCT/PのCGTPと、生物製剤としてのcGMPを組み合わせる構造 、EUはATMPを医薬品の一類型として扱い、ATMP専用GMPガイドラインで管理する構造 になっている点です。
日本・米国・EUにおける再生医療等製品関連GxPの比較表
項目 日本 米国 EU 主な製品分類 再生医療等製品 HCT/P、細胞・遺伝子治療製品、生物製剤 ATMP:Advanced Therapy Medicinal Product 法制度上の考え方 薬機法上、医薬品・医療機器とは別に「再生医療等製品」として区分 361 HCT/Pと351 HCT/P・生物製剤で規制の重さが異なる ATMPは医薬品規制の枠内にある特殊な製品群 製造所の主なGxP GCTP省令 CGTP:21 CFR Part 1271、加えて生物製剤ではcGMP等 EU GMP、特にATMP専用GMPガイドライン GMPとの関係 医薬品GMPではなく、再生医療等製品にはGCTPが主基準 HCT/PではCGTP、治療目的の細胞・遺伝子治療製品ではcGMP・生物製剤規制も関係 GMPの中にATMP専用の枠組みがある 製造販売業者・承認保有者の品質管理 GQP省令が適用 日本型の「GQP省令」はない。BLA/IND、cGMP、品質システム、FDA査察等で管理 MAH、QP、GMP、PQS、バリエーション管理等で管理 市販後安全管理 GVP省令 FDAの有害事象報告、REMS、postmarketing requirements等 EU GVP、RMP、PSUR/PBRER、ATMP特有の安全性・有効性フォローアップ 製造販売後調査 GPSP省令、使用成績調査、条件及び期限付承認後の評価 postmarketing studies/requirements、real-world evidence、accelerated approval後の確認試験等 PASS、PAES、RMP、ATMPの安全性・有効性フォローアップ 条件付き・早期承認制度 条件及び期限付承認制度 RMAT、accelerated approval等。ただしRMATは承認そのものではなく開発促進指定 Conditional marketing authorisation、approval under exceptional circumstances、PRIME等 医療機関内での例外的使用 薬機法上の製品とは別に、再生医療等安全性確保法の枠組みがある 361 HCT/Pでは市販前承認不要の場合があるが、要件を外れると351製品として規制 Hospital Exemptionがあるが、加盟国管理かつATMP特有の例外制度 特徴的な違い GCTP・GQP・GVP・GPSPが比較的明確に分かれる HCT/P、biologics、drug/device combinationの分類で適用規制が大きく変わる ATMPを医薬品GMP体系の中で扱い、ATMP専用GMPとRMPを重視
PMDAは、日本の再生医療等製品について、薬機法上の製品区分として説明しており、GMP/QMS/GCTP適合性調査では、医薬品等はGMP、医療機器等はQMS、再生医療等製品はGCTPという形で製造所管理基準が整理されています。
製造管理・品質管理GxPの比較
項目 日本:GCTP 米国:CGTP / cGMP EU:ATMP GMP 主な名称 GCTP CGTP、cGMP GMP specific to ATMPs 主な対象 再生医療等製品の製造所 HCT/P製造施設、細胞・遺伝子治療製品製造施設、生物製剤製造施設 ATMP製造施設 法的位置づけ 再生医療等製品専用の省令 21 CFR Part 1271のCGTP、必要に応じて21 CFR 210/211、600番台、生物製剤規制 EudraLex Volume 4のATMP専用GMPガイドライン 管理の中心 製造管理、品質管理、無菌性、ドナー管理、トレーサビリティ HCT/Pでは感染症伝播防止、ドナー適格性、汚染防止。治療製品ではcGMP・CMCが重要 ATMPの特性に合わせたGMP、リスクベース管理、QPによるバッチ認証 日本との大きな違い GCTPという独立した専用省令がある 「再生医療等製品GCTP」に相当する単一制度ではなく、HCT/P分類と生物製剤規制の組合せ GCTPという名称ではなく、GMPのATMP専用枠組みとして整理
米国では、HCT/Pに対して21 CFR Part 1271のCurrent Good Tissue Practice、いわゆるCGTPが定められています。CGTPは、HCT/Pによる感染症の導入・伝播・拡散を防ぐことを目的に、ドナー適格性、汚染防止、製造時の管理などを求めるものです。
EUでは、ATMPに特化したGMPガイドラインがEuropean Commissionから公表されており、ATMPの特性に合わせたGMPとして運用されます。
GQP相当の品質保証体制の比較
項目 日本 米国 EU 名称 GQP 日本のGQPに相当する独立名称は通常使わない 日本のGQPに相当する独立名称は通常使わない 主体 製造販売業者 Sponsor、Applicant、BLA holder、manufacturer等 MAH、Manufacturer、Qualified Person等 主な機能 市場出荷、製造業者管理、品質情報、品質不良、回収 CMC、cGMP、FDA査察、BLA/IND管理、品質システム、回収等 GMP、PQS、QP release、MAH責任、variation管理、recall等 日本との違い GQP省令として明確に制度化 品質保証機能はあるが、「GQP省令」という独立カテゴリーではない MAH/QP/GMP/PQSの組合せで品質保証機能を担う
日本では、再生医療等製品にもGQP省令が適用され、製造販売業者の品質管理体制が明確に求められます。
市販後安全管理・市販後調査の比較
項目 日本 米国 EU 安全管理GxP GVP省令 FDAの有害事象報告、REMS、postmarketing requirements等 EU GVP 市販後調査 GPSP省令 postmarketing study、postmarketing requirement、real-world evidence等 PASS、PAES、RMP 再生医療等製品で重視される点 条件及び期限付承認後の使用成績評価、安全性情報収集 長期フォローアップ、遅発性リスク、確認試験、RWE RMP、安全性・有効性フォローアップ、長期追跡 特徴 GVPとGPSPが制度上分かれている 製品特性・承認経路に応じて市販後要件が設定される ATMP特有の安全性・有効性フォローアップガイドラインがある
EUでは、ATMPについて、通常のGVPに加えて、安全性・有効性フォローアップとリスクマネジメントに関するATMP特有のガイドラインがあります。このガイドラインは、ATMPの承認後の有効性・副作用フォローアップ、リスク管理に関する考え方を扱います。
早期承認・条件付き制度の比較
項目 日本 米国 EU 主な制度 条件及び期限付承認 RMAT、accelerated approval、breakthrough等 Conditional Marketing Authorisation、PRIME、approval under exceptional circumstances等 再生医療等製品との関係 再生医療等製品で特徴的に使われる制度 RMATは再生医療治療の開発促進指定。承認そのものではない ATMPにも条件付き承認、PRIME等が関係し得る 承認後の重要点 使用成績評価、追加データ、通常承認・再審査への対応 確認試験、RWE、安全性追跡、postmarketing commitment RMP、PASS/PAES、追加データ、risk minimisation 注意点 「早期に販売できる制度」だけでなく、承認後評価が重要 RMAT指定は開発促進であり、承認基準を下げるものではない Hospital Exemptionと中央承認ATMPを混同しない
米国のRMAT指定は、IND提出時またはIND amendmentとして申請する開発促進制度であり、承認そのものではありません。
日本・米国・EUの違いを一言で整理
地域 一言でいうと 日本 再生医療等製品を独立した製品区分として扱い、製造所はGCTP、製造販売業者はGQP/GVP/GPSPで整理する 米国 HCT/Pか、生物製剤・細胞遺伝子治療製品かで規制経路が大きく変わり、CGTPとcGMPの組合せで考える EU ATMPを医薬品規制の枠内で扱い、ATMP専用GMP、MAH/QP、RMP、GVPで管理する
注意点・例外
米国では、同じ細胞・組織由来製品でも、361 HCT/Pとして扱われるか、351 HCT/P・生物製剤として扱われるかで規制要件が大きく変わります。EUでは、中央承認ATMPとHospital Exemptionを混同しないことが重要です。日本でも、薬機法上の再生医療等製品と、再生医療等安全性確保法に基づく医療機関での再生医療は別制度です。
個別製品の国際開発・申請戦略では、分類判断、CMC、GMP/GCTP/CGTP/ATMP GMP、GCP、GLP、GVP、市販後調査、長期フォローアップの扱いが製品ごとに変わります。専門家に確認が必要です。
参考文献・出典
【確実性: 高】
![CTDはどのような医薬品関連手続きに使われるのか (NDA,BLA,MAA,IND,IMPD,MF,DMF,BMF,ASMF)[2026/06/06]](https://harikiri.diskstation.me/wp-content/uploads/2021/02/80F3D755-3019-49C6-BDAE-47D137C87826.jpeg)
![バイオ医薬: 造船DXから考える大型バイオ医薬品製造設備の設計 [2026/05/31]](https://harikiri.diskstation.me/wp-content/uploads/2026/05/image-12-2.jpg)