カテゴリー: BIOLOGICS
-
[用語] 4型コラーゲン= Type-IV collagen [Biotech] [2022/09/03]
Post Views: 826 4型コラーゲン= Type-IV collagen 網目構造をとる繊維性コラー…
投稿者

-
[用語] RNA, tRNA, rRNA, etc. [Biotech] [2022/09/03]
Post Views: 884 RNA; ribonucleic acid DNAの構成要素は,アデニン(A)…
投稿者
-
[用語] rAAVとは.遺伝子治療用に遺伝子組換えされたAAVである [2025/04/21]
Post Views: 854 rAAV recombinant AAV (Adeno Associated …
投稿者
-
[用語] Protein A; プロテイン A [Bio-Tech] [2022/09/03]
Post Views: 839 Protein A Protein Aは、Staphylococcus aur…
投稿者
-
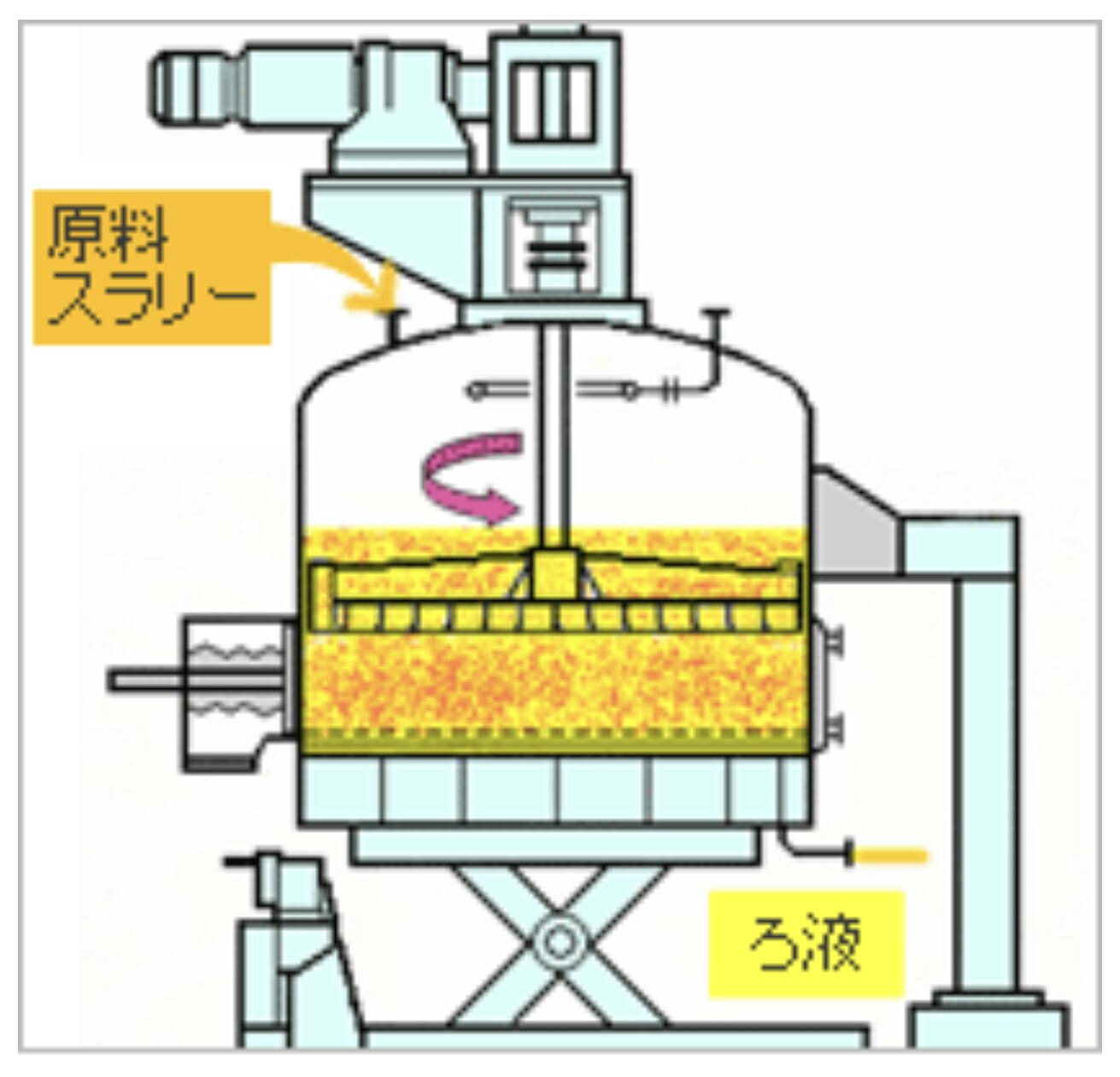
[Bio-Equip] 沈澱化した目的物をろ過により脱水し乾燥させてパウダーを得る – 加圧濾過器/ 日本化学機器製造株式会社 [2022/05/03]*
Post Views: 830 はじめに バイオ医薬品をつくるには,種々の高価な装置が必要です.MRHARIK…
投稿者

-
[Bio-Equip] ホモジナイザーは細胞の破砕処理装置 / 連続処理には圧力式のFrench Press (GEA社)、バッチ処理にはビーズを用いたBead Mill (ELE) / その他原理を紹介
Post Views: 879 はじめに バイオ医薬品をつくるには,種々の高価な装置が必要です.MRHARIK…
投稿者

-
[Bio-Equip] リアルタイム緩衝液作成装置 – Allegro Connect buffer management system (BMS) [2020/12/16]*
Post Views: 908 はじめに バイオ医薬品をつくるには,種々の高価な装置が必要です.MRHARIK…
投稿者