タグ: API
-
[用語] Safety; 医薬品の製造から出荷における安全性 – どのように確保されているのか – GMP Compliance [2023/10/15]
Post Views: 1,122 はじめに 医薬品は、GMPというルールに従い、予め定めた方法に従い作業を実…
投稿者

-
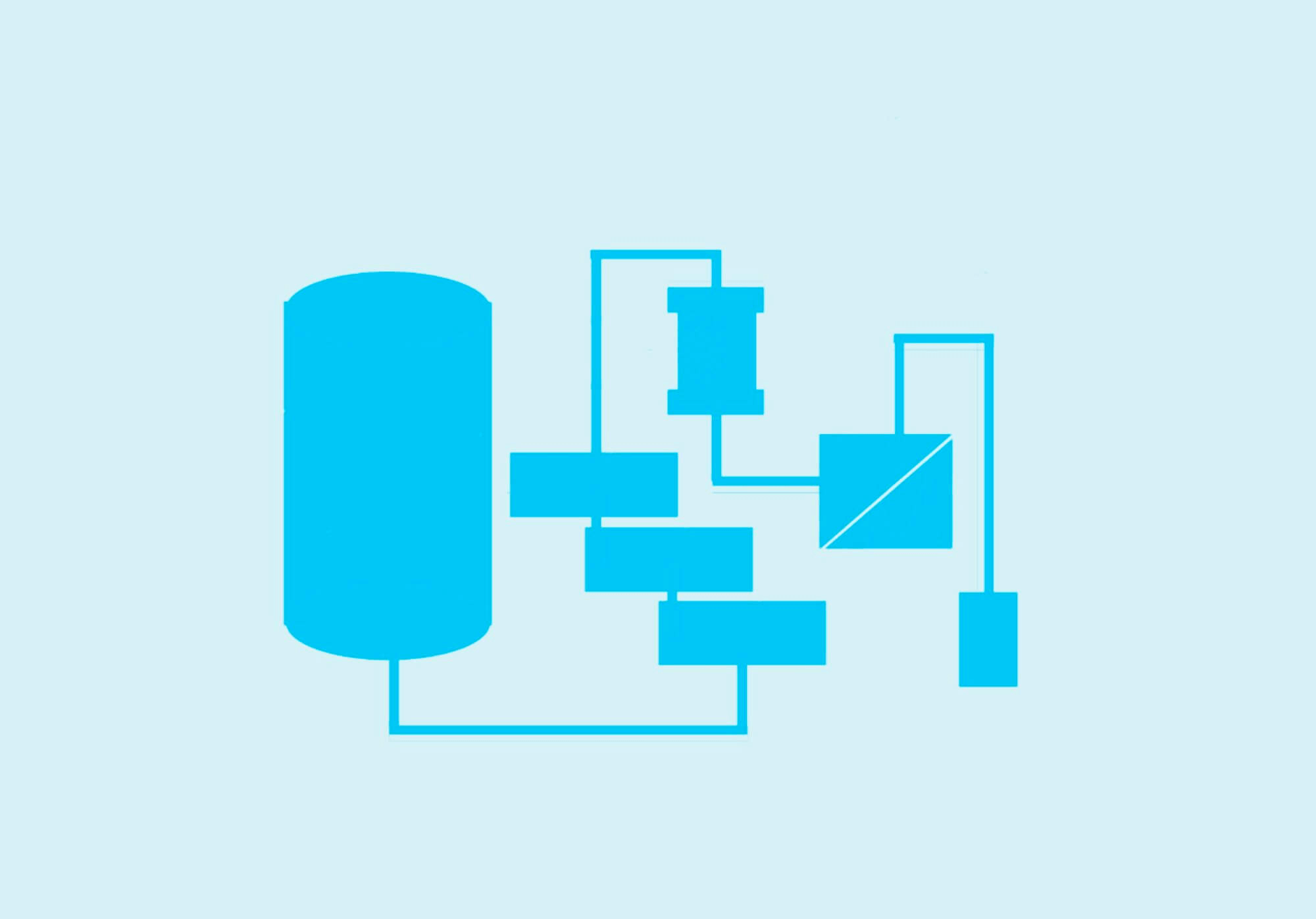
[Biologics] 医薬品製造における工程別の名称 : 原料からAPI(有効成分) / DS (原薬),バイオ医薬と低分子医薬での比較 [2025/04/10改定]
Post Views: 1,415 工程別の名称 医薬品の製造は、原料(raw material)を使い、途中…
投稿者
-
[用語] API ; active pharmaceutical ingredient
Post Views: 861 API: 有効成分(API:active pharmaceutical ing…
投稿者