Post Views: 271
はじめに
WordPressは,PHP + MySQL (mariaDB)ベースの動的なCMSです.DSMに内蔵されたWeb Station (ApacheまたはNginx) + PHPがそれぞれ処理します.
今回,一つのNASの上に2つ(以上)のサイトを構築する手順を解説します.
以前にAI君にその方法を問うてみると「できる」と断言されました.その詳細な方法について色々と聞いてみると「それらしい答え」を出してきました.
これまでは,ネット検索では思いついた時に,その都度調査していましたが,求めているサイト/方法/手順は見つけられませんでした.あきられて新しいIP Addressを取得(費用が必要)することも真剣に考えていところです.
しかし,1カ月前,AI君は「できる」と断言しました.その後,1カ月くらい放置して自分の頭の中で熟成させていたこの案件ですが,この土日,及び+3日を使い,更にその詳細についてAI君と共に検討した結果,1台のSynology NASの上に2つのサイトを構築し安定稼働させることができました.2つ目以降のサイトの内容はこれから作っていこうと思ています.
作業概要は,
(1) DSM->File Stationで既存wordpressフォルダー複製
(2)phpMyAdminによる既存のData Baseの移行と修正
(3) Web Stationによるportal(仮想ドメイン?)の設定,
(4) DMSでのText Editorによるwp-config.php, .htaccessの修正,
等です.
当初の検討では,Conatainer Managerを使用したwordpressのコンテナ化によるマルチドメインを検討しましたが,結局は,Web StationのVirtual Host(仮想ホスト)を使用することで一つのNASの上に複数のサイト構築が可能でした.
以下に詳細を解説します.なお,Synology NASの解説は,日本語版では冗長なのでそれを避けて用語などは英語版の表記としています.
構築後しばらくの間で稼働状況についてモニターリングし,追加の対策を実施している.具体的には,cache関係,portal severの設定関係で,トリッキーな挙動(ここに気づくのに時間かがかかった)と安定稼働には必須の設定を追加した.
構築方法
作業する前にバックアップを取ってから進めてるのが良いです.
作業前のバックアップ
hyper backupで現時点での/web以下(既存のwordpressファイルが格納されているfoler)をバックアップします.その他,以下の設定で実行しておきます.
設定
Folder : /volume1/web
Application : MariaDB 10
Web Station
Backj up nowボタンを押す
参考 : [Synology] NAS (DS918+/DS920+)の バックアップを考える …
DDNSの取得
以下の方法により構築する2つ目のDomain Nameを用意する.
Synology NASの場合,2台以上のNASを持っているのであれば,External Access->DDNSからSynologyからDomain NameをNASの1台に付き1つの名前を頂ける.
myDNS.jp様のサイトから複数個のDomain Nameを頂ける.
Let’s Encription証明書
証明書については,Synology NASを複数持っているので,それぞれについてSynologyからDomain Nameを取得している.
表1. Let’s Encrypt の代表的なレート制限
制限の種類 内容 同一FQDNでの再発行(同一証明書内容) 1週間に5回まで 同一登録ドメイン(例:example.com)に対する新規証明書発行 1週間に50回まで 失敗した認証試行数 1時間に5回まで アカウントごとの登録レート 制限あり(通常の使用で問題なし)
Web Stationでの設定
設定の前準備(Control Panelでの作業で示している)として以下のAccess Control Profileを作成しておく.これは安全装置であり、即使用するものではなく、メンテナンス時に有効化して外部からのアクセスを完全に遮断する装置となる。
Longin Portal->Advanedを開き、以下の設定を作成する.
192.168.11.0/24=Allow、All=Denyとしているのは、192.168.11.0に対してLocalからのアクセスのみを許可する設定とする。これは、IPレベルにる入口制御である。
具体的な機能は以下の通り。
Webアプリ(wordpress)の前段での制御である。即ち、防御の最前線である。
認証画面すら表示させない
アプリ脆弱性以前に”通信自体”を止める
表2. Access Control Profile
前準備は完了した。
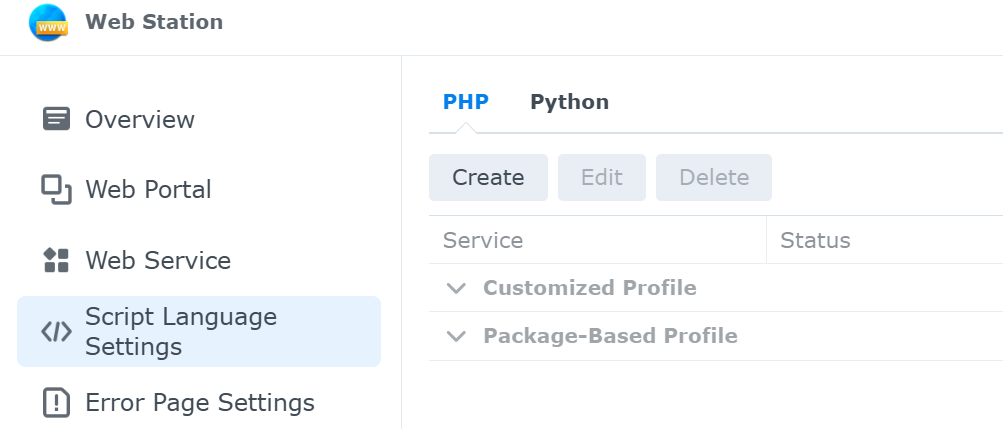
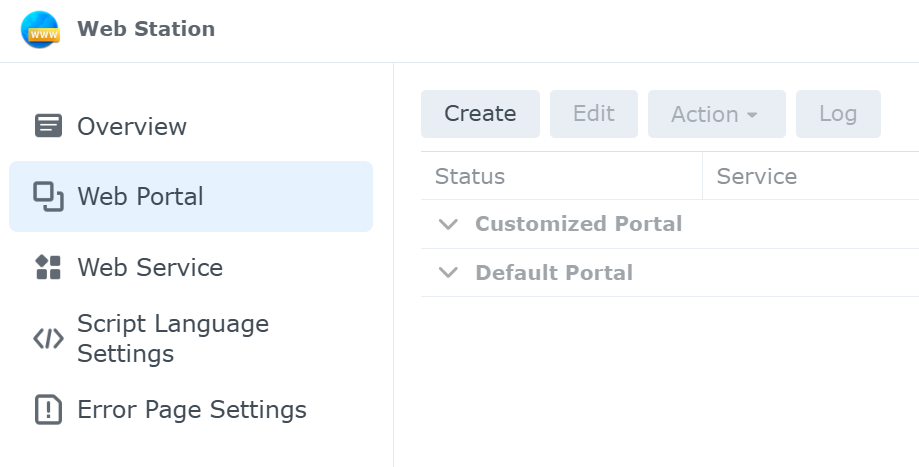
web stationでは,以下の項目を設定することができる.web stationでの設定手順は,(3) Script language Settings →(2) Web Service →(1) Web Portalの順となる.
Web Portal
Web Service
Script Language Settings
Error Page Settings
図1. Web Stationの設定画面 (Script Language Settingsを選択している状況)
Script Language Settingsでprofileを作成:
作業
Creatボタンを押す
Create Profile – Configre general settings画面が現れる
以下の項目に入力する(後で修正は可能)
Profile name : <profile for 2nd site>
Description : <同上など,適当に>
PHP version : <インストールしているphpのバージョンがリストされるので,一つを選ぶ,ex: php8.2>
Enabe PHP cache : チェックする
Enable Xdebug : チェックする
Enable display_erros to display PHP error message :チェックする
(以上のチェックは,作成後に変更可能)
Customized PHP open_basedir : Default
Nextボタンをクリック
Configure extensions画面が現れる
全てを選択(理解できていないので全て選択,重複エラーでは重複項目のチェックを外す)
Nextボタンを押す
Configure FPM settings画面が現れる(FastCGI Process Manager)
FPM mode : dynamic (PHP-FPMを使用せず,web serverがPHPを直接処理する)
Max processes: 102 (default)
Start servers : 2 (default)
Min spare servers : 2 (default)
Max spare servers : 4 (default)
Nextボタンを押す
Configure core settings画面が現れる
mysqli.default_socket : /run/mysqld/mysqld10.sock (に修正, wp-config.phpの記載に合わせる)
pdo_mysql.default_socket : /runmysqld/mysqld10.sock (に修正, wp-config.phpの記載に合わせる)
残りは規定値のまま
Nextボタンを押す
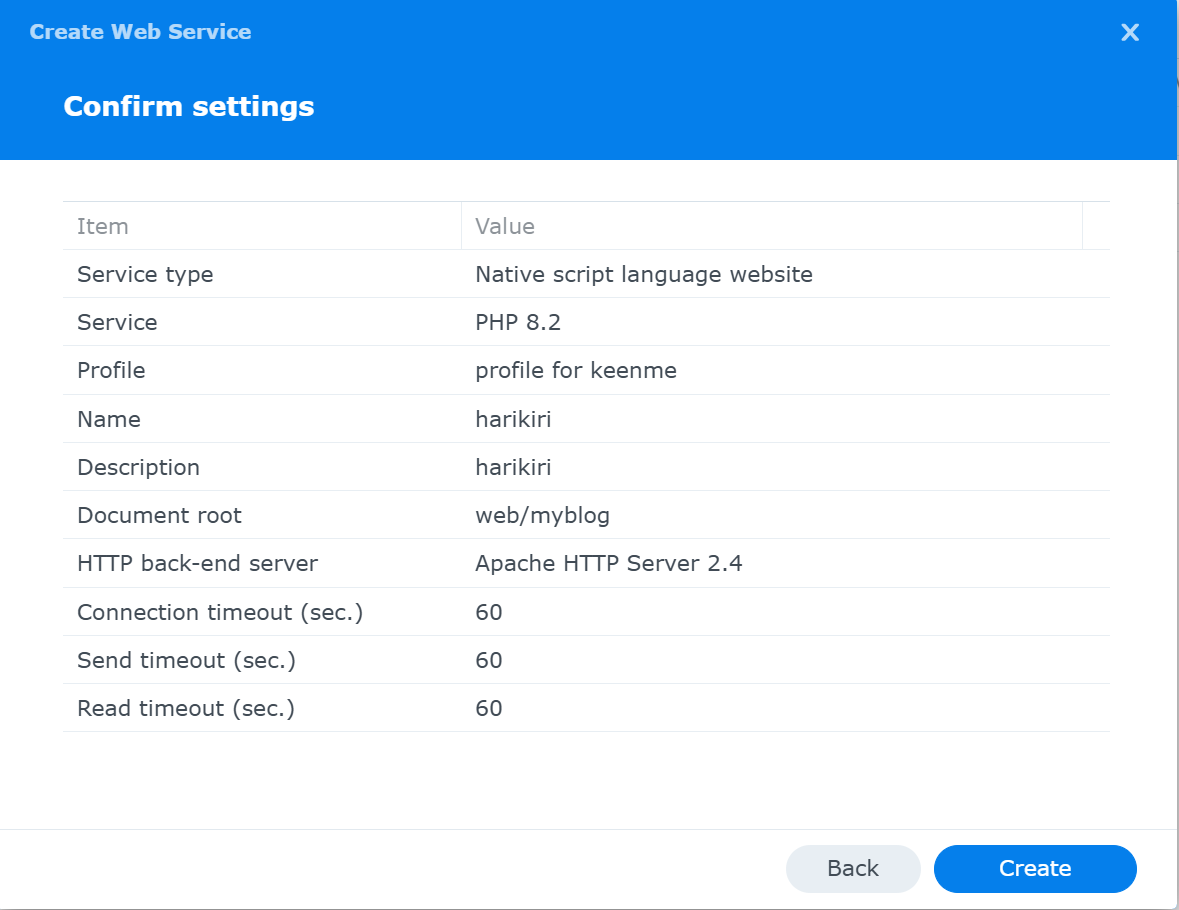
Confirm settings画面が現れる
以上
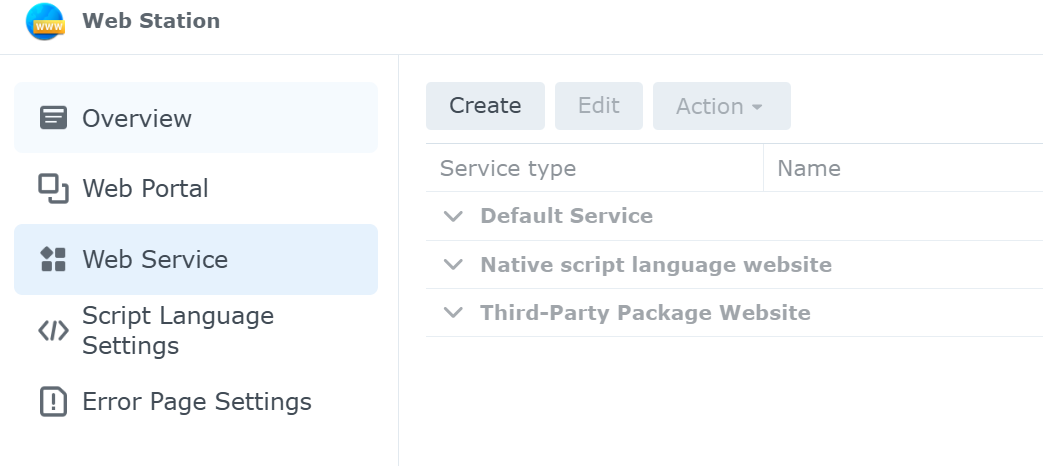
Web Service の作成:
Web Serviceでは,以下の項目が表示される.
Default Service
Native script language website
Third-party Package website
図2. Web Stationの設定画面 (Web Seviceを選択している状況)
今回の作業では,Native script language websiteを作成する.
Web Serviceは,site Aとsite Bで共用は可能だが,今回は,専用にそれぞれ作成した.
作業:
Createボタンを押す
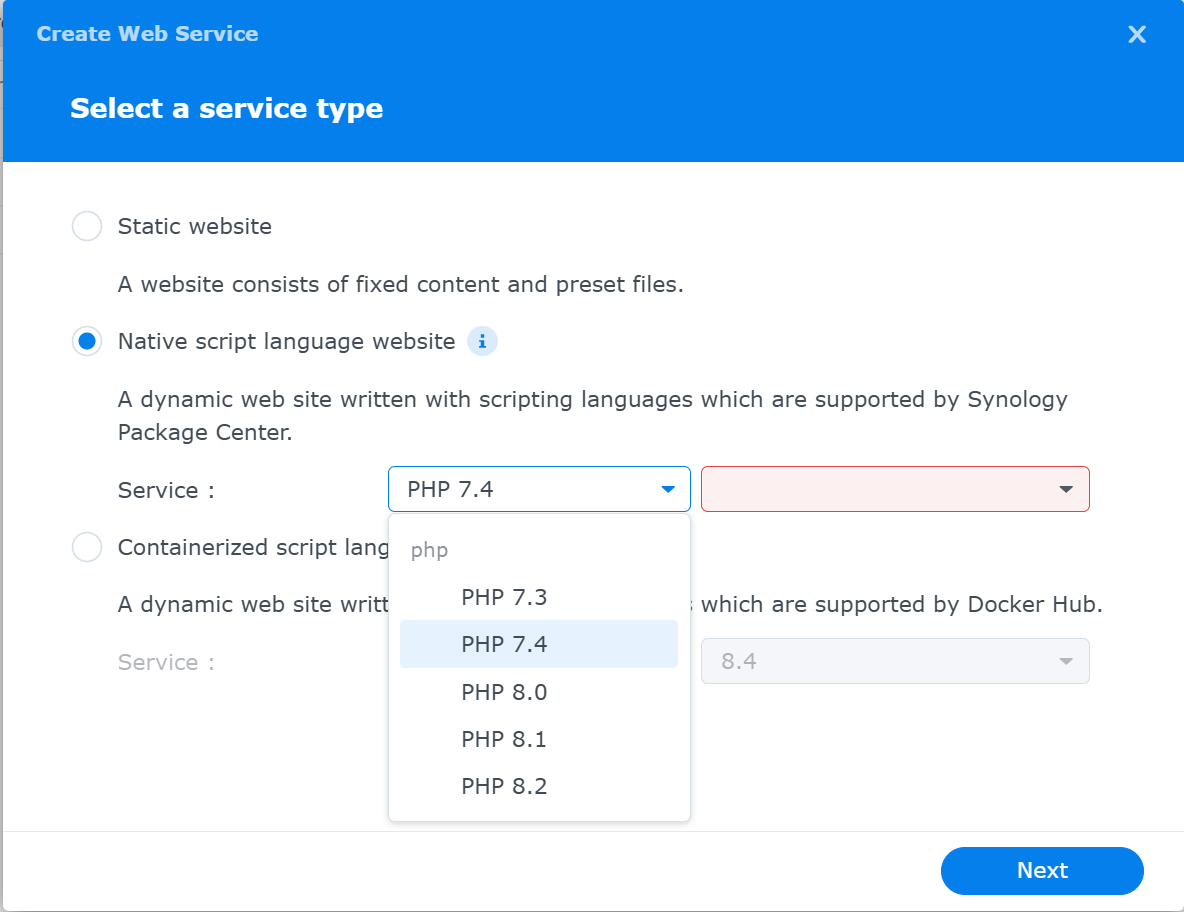
Select a service type画面が現れる
static websiteとContainerized script language websiteは選択しない.
Native script language websiteをチェック(選択)する
Service : リスト他されphpの中から「script language settings」にて作成した時に設定したphp version である「php 8.2」を選択する
右隣のプルダウンメニューには,「script language settings」で作成したprofileがリストされてるのでそれを選択する.
Nextボタンを押す
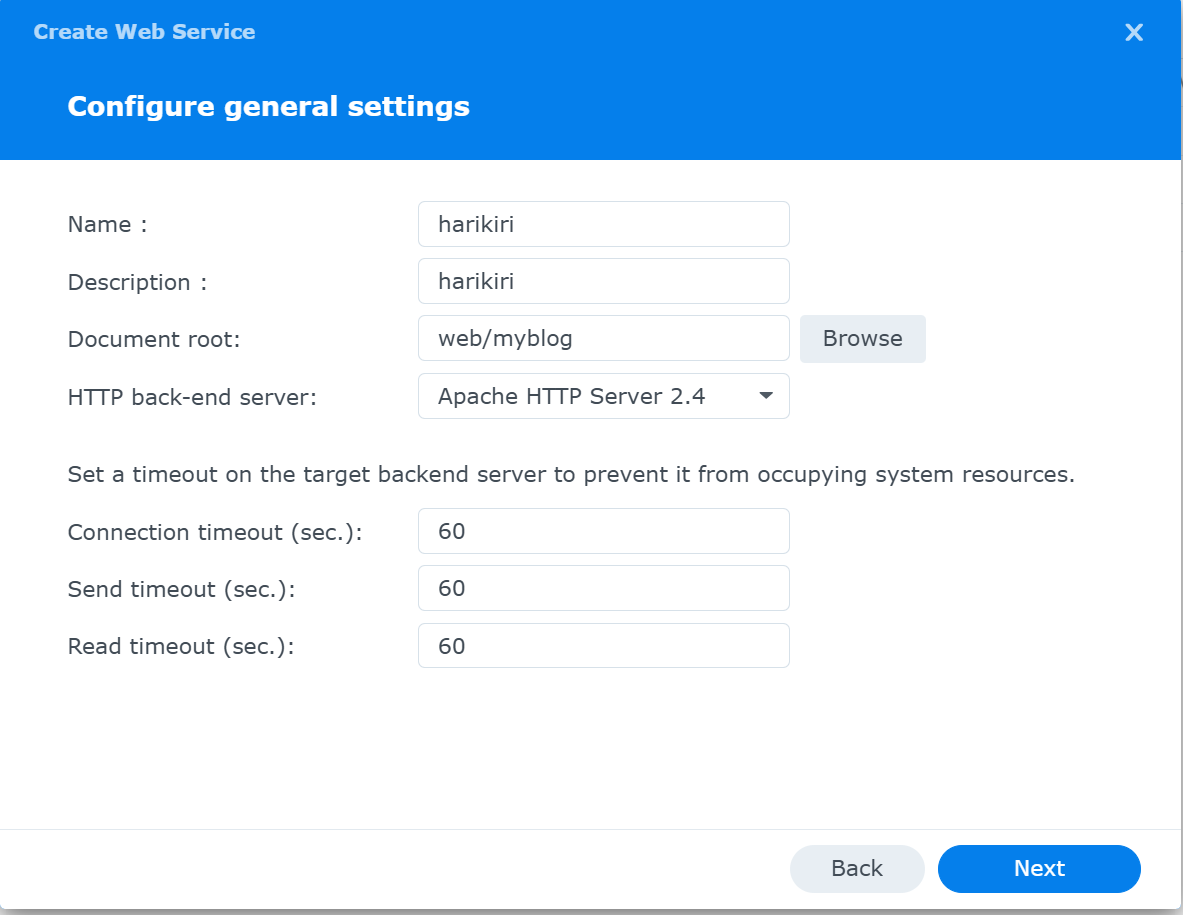
Configure geral setting画面が現れる
Name : <site B用であることが分かるような名前にする>
Descriptiion : <適当に>
Document root : /web/<site B>
HTTP back-end server : Apache HTTP Sever 2.4
残りの項目は規定値(default)
Nextボタンを押す
Confirm settings画面が現れる
Createボタンを押す
Virtual hostを作るには,http groupのread permissionが必要であり自動で作成するとのメッセージが出るので,「OK」ボタンを押す
Native script laguage website項目に,Service typeはPHP,Nameには作成した名前でリストされる.
完了
以上
Web Portalの作成 :
web Portalでは,以下の項目が表示される.
Customized Portal
Default Portal
図3. Web Portal 画面
今回の作業では,Customized Portalを以下の手順で作成する.
図4. Web Profile設定
作業:
Createボタンを押す
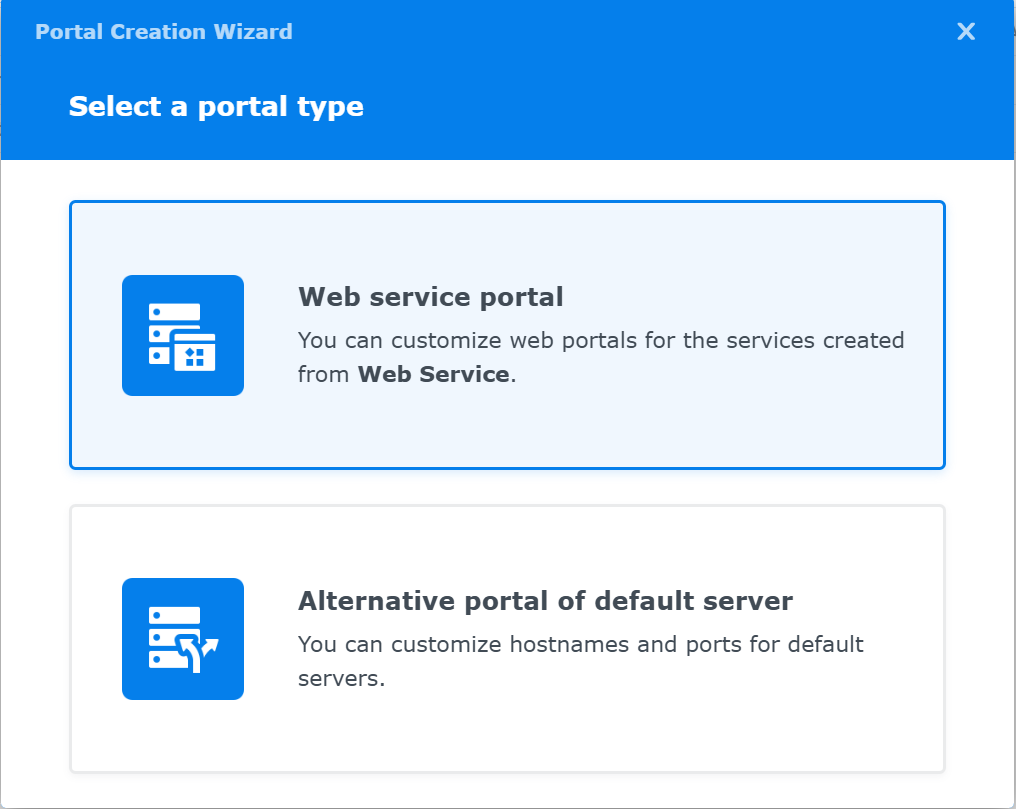
Select a portal type画面が現れる
Web service portalをマウスクリックする.
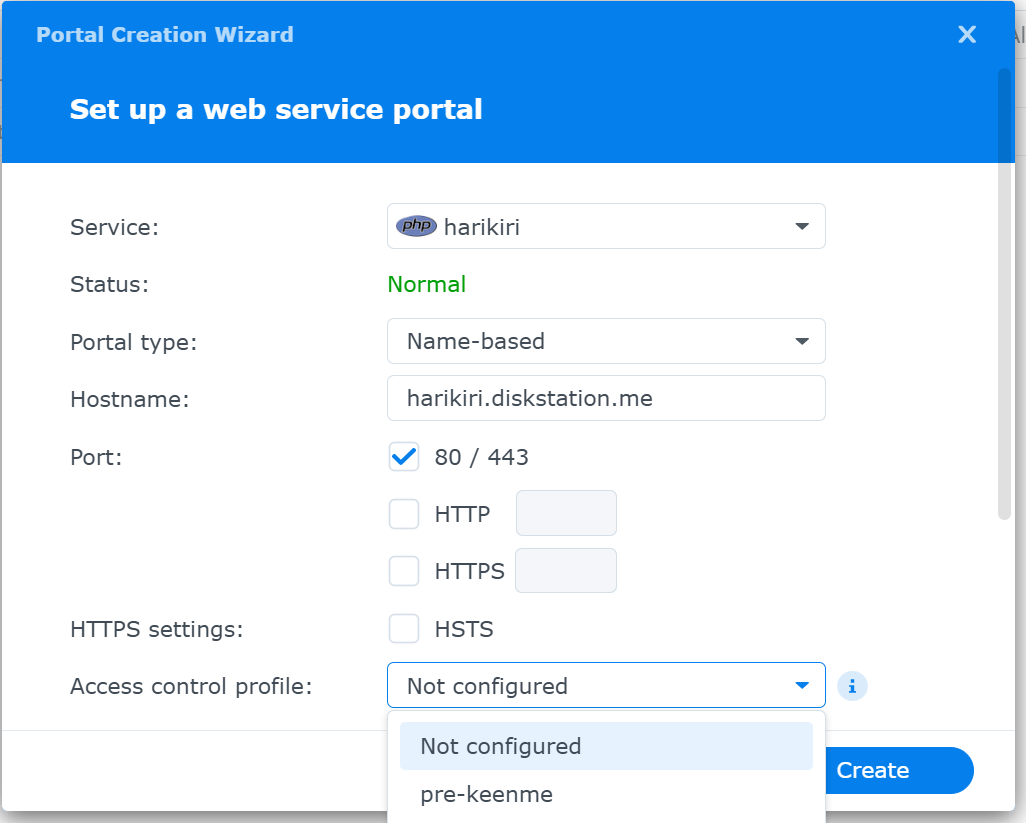
Set up a web service portal画面が現れる
Service : <作成したweb serviceがリストされるので,それを選択する>
Status : Normal (選択したweb serviceが正常な場合にNormalと表示されている)
Portal type : <Name-based
Hostname : <domain name>, ex:????.myDNS.jp
Port : 80/443 (でもOKだと思う), 今回はHTTPS: 443とした
HTTPS settigs : HSTSをチェック
Access control profile : <Access Control Profileの作成で作成したプロファイル名をリストから選択する.
Error page profile : Default error page profile (defaultのまま)
Enambe access log : 今回はチェックなし
Createボタンを押す
Customized portal項目に作ったPortalがリストされる
完了
以上
注) Synology NASのDSMにあるDDNSは、1つしか登録できない。従って、DDNSプロバイダ(myDNS.jp)には2つ目のurlは登録できない。当サイトでは、synology製品に付いてくるドメインを1つ目のurlに設定している。
複数サーバー安定化に忘れてはいけない
一番の不安定原因は,cache pluginのコンフリクトが考えられる.その他,portal serverの設定がある.
redisと一般的なwordpressのcache pluginはキャッシャ層が異なる.レスポンス速度を高めるために一般的なキャッシュ(表示系)とredisを導入しているので,以下の事項について注意して進める必要があった.
一般的なキャッシュプラグイン
サイト構築の設定過程では,個々のwordpressのcache pluginの挙動を意識して進める必要がある.また,作業中はcache pluginは無効にしておくか,そうでない場合は適時キャッシュをflushする.
設定が完了してから無効化していたcache pluginを有効化する.
redis関連 pluginの設定
以下に解説しているweb stationでPortal Serverを同時にenableにする場合には注意が必要である.
先ず問題だった点は、
今回の場合,Redis Serverを複数のwordpressを導入した1つの同じNAS内にcontainer managerでコンテナ構築(host)し,更に、個々のwordpressでは、それぞれredis object server pluginをインストールしていた.このpluginでの設定ではdefaultのredis 用のDBの番号は「0」となっていたため、複数のportal(web url)をenableにした途端にredisのキャッシャでコンフリクトを起こした.
対処:
redis serverに関わる以下のコードをwp-config.phpに追記する.
define( 'WP_HOME', 'https://harikiri.diskstation.me' );
define( 'WP_SITEURL', 'https://harikiri.diskstation.me' );
define( 'WP_CACHE', true );
define( 'WP_REDIS_HOST', '127.0.0.1' );
define( 'WP_REDIS_PORT', 6379 );
define( 'WP_REDIS_DATABASE', <0,1,2・・・> );
define( 'WP_CACHE_KEY_SALT', '<site name>_ ' );note :
更に必要な作業
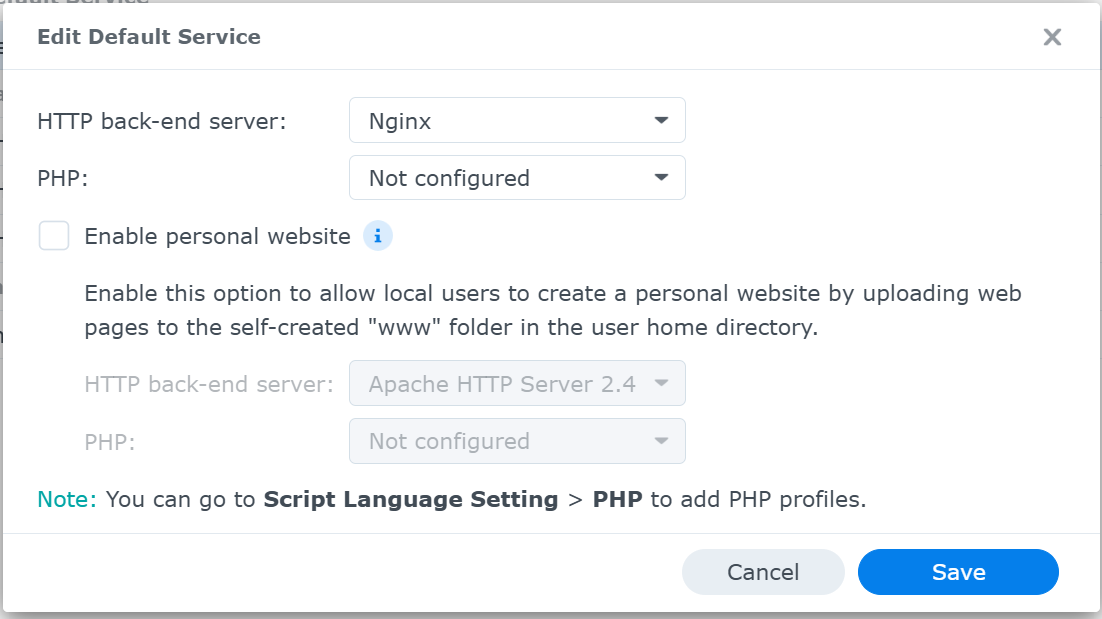
default serverの設定
Web Protalで複数のweb serverを安定的に稼働させるために,default serverの設定をいじる必要がある.なぜなら,Multi Domain Web Serverを構築する前の状態はSingle Web Server (既存)が稼働するのみであり,それはdefault server が担っていたため.
今回の作業過程では,既存のweb serverを新しくportal server (以降で説明)として設定 (既存のurl:harikirii.diskstation.me)したため,既存のurlであったdefault server (url: 同じ)が起動すると,この同じurlに対して異なるservice (script languageを含む)が割り当てられる ことになるため,コンフリクトを生じる可能性が考えられた.
そこで,以下の手順により,default serverに対して適切な設定が可能になるようservice設定をいじった.この設定を完了させてから,複数(現在3つ)あるportal(web server)がレスポンスを失う自体にはなっていない(2025/06/20).
設定 :
先ず,Web Portal 設定の画面では,default serverをdisable/enableは,できな仕様となっている(図5).
そこで,Service設定画面から設定を以下の手順でいじる(図6).
図6に示すように,user portalが使用しているHTTP back-end serverはapacheである.念のため,default serverにはそれとは異なる「Nginx」を選択する.
Serverが起動しないようにすめにたに(結果的そうだった*),PHPには「Not configured」を選択する.おそらくこの設定が重要.
* NASをrebootさせてlogを確認してみると,この設定前ではharikiri.diskstation.meの起動が2回記録されていた.設定してからは,1回の起動のみが記録されるようになった.
Save
以上
図5. Default serverのdisable/enableはできない(仕様)
図6. Service設定画面
Portal Serverに関する設定
今回の作業では,既存のwebサイト(harikiri.diskstation.me)は,Default serverで管理されていた(そのはずな)ので,専用のportalはリスト(登録)されていなかった.
そこで,harikiri.diskstation.me用に上記の作業でWeb Portalを作成した.その際,/web/<site A用のfolfer>のフォルダ構造と,harikiri.diskstation.me/<site A>というurl構造が不整合となりブラウザでの表示が出来なくなった(エラー500, 404).
修正作業は,以降に述べているように「wp-config.php」の修正,DBの複製と修正,及び「.htaccess」の修正が必要.
File Stationから,既存のwordpressディレクトリを複製して2つめwordpressディレクトリとし,wp-config.php,.htaccess,必要があればrobots.txtを修正する.
以下詳細を示す.
File Stationでの作業
(1) DSM->File Stationで既存のwordpressフォルダーの複製
/web/<site A>にwordpressがインストールされている前提とする(既存).
/web/<site B>をcreate folderで作成する.
<site A> foldeにあるfolder/fileの全てを<site B>に複製する.
(2) .htaccessの修正
以下のコードでにおいて “/” が,”/<site A/Bのfolder name>”になっているとブラウザ表示できないので,以下のコードに置き換える.
# BEGIN WordPress
<IfModule mod_rewrite.c>
RewriteEngine On
RewriteBase /
RewriteRule ^index\.php$ - [L]
RewriteCond %{REQUEST_FILENAME} !-f
RewriteCond %{REQUEST_FILENAME} !-d
RewriteRule . /index.php [L]
</IfModule>
# END WordPress(3) wp-config.phpの修正
MariaDB10のデータベース(DB)は,2つ目のuser2で別DBを作成し,exportしたuser1のDBをuser2のDBにimportされていることを前提にしている.
define( 'DB_NAME', 'DB2' );
define( 'DB_USER', 'user2' );
define( 'DB_PASSWORD', '********' );
define( 'DB_HOST', 'localhost:/run/mysqld/mysqld10.sock' ); // MariaDBパッケージならlocalhostでOK(未確認)
define('DB_CHARSET', 'utf8mb4');
define('DB_COLLATE', '');/volume1/web/<site A filder name>/wp-config.php に以下の2行を require_once より前
define( 'WP_HOME', 'https://harikiri.diskstation.me' );(4) robots.txtの修正
robots.txt (小文字)についてもサイトに合わせて修正してください.
Control Panelでの作業
Login Portal -> Advanced -> Access Control Profileでprofile作成画面を開き当該profilelを作成する.
access control profileは,web stationの設定で使用する.Web Stationの設定が完了後,後から当該profileを作成・設定も可能.
機能:
サイトの内容を編集している間やその他のIP Addressを拒否/許可することが可能.ネットからのアクセスを拒否する設定のprofileを作り,web station->web portal->”で,作成したportalのAccess control profile”に指定するとprofileに従ってアクセス制限される.
サイトの投稿が整いサイトを公開したい場合,公開用のprofileを前述と同様のところに指定するか,何も指定しなければフルアクセスとなる.
access control profileを作る
Login Portal->Advancedを開き,Access Control Profileを開く
Createボタンを押し,2つ目のサイト用にprofileを作成する
MariaDB 10
WordPressはMariaDB 10のデータベース(DB)にページを保存しており,2つ目のサイト構築にも専用のDBが必要である.
今回は,既存のuser1が持っているデータベース(DB1)をphpMyAdminからexportしてDB1をダウンロード,新しいuser2で作成したDB2にimport して,これをSite-2用のデータベースとして修正し稼働できることを確認する.同時にphpMyAdminの使い方の習得を並行して行った.
以下のphpMyAdminで行う操作は,慎重に行ってください.
特に置換を実施する際,先に検索でヒットするテーブルやレコードを確認しておき,その後に,置換の作業をするようにすれば理解しながら進められます.
既存DB1のexport
phpMyAdminにuser1(root権限)でログイン
site-2用のDB1をexportしてsql形式でdownloadしておく
ログアウト
新しいuserとDBの作成
phpMyAdminに既存のuser1(root権限)でログイン
新しくuser2を作成
上にあるタブの”権限”で表示される”新規作成”
ユーザーと同名のデータベース作成、グローバル権限なし
実行
新しくDB2 (文字コードはutf8mb4_unicode_ci)を作成
user2にDBのアクセス権限を与える
DB2をクリックして選択
タブメニューの”権限”クリック
ユーザアカウントを追加するをクリック
UID/PD設定
データベースDB2へすべての権限を与える」をチェック
実行
sqlの場合
GRANT ALL PRIVILEGES ON DB2.* TO user2’@’localhost’;
FLUSH PRIVILEGES;
DB2を選択
メニューのimportを選択
先にexportしたDB1を選択
実行(DB2にDB1(の内容:全ての表)をimportする)
siteurlとhomeのurlの修正
site 2用のDB2をクリック
wp_optionsテーブルをクリック
option_nameカラムにsiteurlか,homeがあるレコードを探す.両方ともに以下の処理をする
option_valueにあるurlを確認する
もしもwordpressがインストールされているfolder名が含まれていれば,そのfolder名 を削除する(web stationで明示しているのでここには必要ない)
項目 説明 siteurlWordPress本体(wp-login.php, wp-admin, プラグインなど)が存在するインストール先URL homeサイト訪問者に表示されるホームURL(TOPページ)
<wp_posts表の修正>
web station -> web service -> Edit Native Script Language Website -> Document rootには、wordpressが置かれているfolderを明示的に指定しているので,DB上で設定する必要はない.
wp_postsには投稿のguidカラムがあり,そこには、”https://harikiri.diskstation.me/myblog/?p=1″のように一意にidが設定されている.このままでもサイトは稼働するが,site 2の投稿idではないので,site 2用に修正する。投稿数が多いがphpmyadminの置換機能で正しい値に一括置き換えを行う.
site 2で使用しているDB2をクリック
DBの横にある「+」をクリック
全てのテーブルが表示される
テーブルwp_postsをクリックしてレコードを表示させる
タブメニューの「検索」をクリック
表示された「検索と置換」ボタンをクリック
検索条件に,”/site 1のurl/<folder名>”を入力する
置換文字列に,”site 2のurl”を入力する
(注: folder名は必要ない)
カラムに,”guid”を選ぶ
実行
置換する投稿のリストが表示される
問題なければ,”置換”ボタンを押す
以上
挿入内部リンクの修正
page(固定ページ)およびpost(投稿)には内部画像のリンクがあるとむ思う.もしも,ベージの編集画面でリンクが外れてしまった場合は,リンクのアドレスの修正が必要になる.
編集画面でpostを開いた時に張り付けていた画像が表示されない場合はリンクが切れている
原因の一つは,旧サイトのsiteurl : aaa.bbb.ccc/ddd, 及びhome : aaa.bbb.ccc,新サイトのsiteurlとhomeを同一にした場合,投稿内で張り付けていた画像のリンクがきれてしまう.
原因は,画像のあるurlにはdddを含んだリンクとなっているはずで,編集画面のコードエディターに切り替えて確認することができる.
対処 :
以下sqlコードをphpMyAdminにログインしサイトDBに対して実行する.実行前にはDBをexportしておく.その前に事前に対象となるレコードを確認しておく.
事前確認: 旧サイトでの画像が保管されていたリンク存在検査(sqlコード)
SELECT ID, post_title
FROM wp_posts
WHERE post_content LIKE '%/myblog/wp-content/uploads/%';置換 : sqlコードの実行により置換する
旧ディレクトリリンク(%https://aaa.bbb.ccc/ddd%)を含むコンテントを取り出し(where),
https://aaa.bbb.ccc/dddを
https://aaa.bbb.cccに置換する.
UPDATE wp_posts
SET post_content = REPLACE(post_content, 'https://aaa.bbb.ccc/ddd', 'https://aaa.bbb.ccc')
WHERE post_content LIKE '%https://aaa.bbb.ccc/ddd%';
#具体的なコード
UPDATE wp_posts
SET post_content = REPLACE(post_content, 'https://harikiri.diskstation.me/myblog/wp-content/uploads/', 'https://harikiri.diskstation.me/wp-content/uploads/')
WHERE post_content LIKE '%/myblog/wp-content/uploads/%';note : "%" の意味(LIKE句におけるワイルドカード)
書き方 意味 %文字列%文字列を含む (前後に任意の文字列があってもOK) 文字列%文字列で始まる %文字列文字列で終わる
その他の表
その他の表にもwordpressに導入しているpluginなどによっては,前述の処理が必要な場合があると考えられる.
必要に応じて確認して修正する.
まとめ
前提1: 既存のWordPressはSynology PackageではなくWordPressからダウンロードして導入している.
前提2: MariaDB 10はSynology Packageから導入している.
前提3: phpMyAdminはSynology Packageから導入している.
DBは既存からexportし異なるuser権限(user2)でDB2を作りimportした.
ディレクトリ分離: 複製 (copy)して/web/<site 2用のフォルダ>に2つめのwordpress 用とした.それぞれは,独自にブラウザで表示と管理ができるMulti Domainにすることができた.
harikir.diskstation.me(当サイト)用にportalを作りenable (又は作成直後)にすると,ブラウザは,コード500のエラー(internal sever error)や404エラー(ファイルがない)を吐いたが,wp-config.phpや.htaccessの修正で解決できた.
wp-config.phpの修正
データベースに関する環境変数の修正
/web/<site ?用のフォルダ>と,urlでは<site A/B用のwordpressフォルダ名を使用しないので齟齬がある.この修正が必要だった.
RT6600ax Routerでの設定でポート転送していても,web stationの設定により問題なく2つのサイト(一つのNAS上の2つのwordpress)に振り分けられる.ホスト名(ドメイン名)は,やり取りされるHTTPリクエストのヘッダーに含まれるためドメイン名で振り分けされている(ようだ).
検討の過程で,reverse proxyによるport設定も試したが,web stationでの設定しているため,二重の設定となるためreverse proxyの設定は必要でない.
既存のsite 1のみで稼働させていた時は,default serverが使用されていた(と考えられる).今回の検討では,明示的に管理したかったので,2つのサイトにそれぞれCustomized Portal (仮想host / server)を構築した.
AI君が言うには,default severは,定義された任意のドメインに一致しないリクエストが来た場合に応答する「最後の受け皿」となる仮想hostとのこと.
web stationにあるこのdefault serverは削除することはできない.
今回構築したこの「複数サイト構成」の呼び方:(by 4o)
呼び方 対応 解説 マルチドメイン(Multi-Domain) ✅ 適切 異なるドメイン名(FQDN)で異なるWebサイトを運用する構成。harikiri.diskstation.me と もう一つのurl がそれぞれ独立して動作していれば、これは正しくマルチドメイン構成です。 マルチサイト(WordPress Multisite) ❌ 非該当 WordPressの1インスタンス内で複数のサイト(サブディレクトリやサブドメイン)を運用する機能。 シングルサイト ×2(独立サイト) ✅ 技術的にはこちらも正しい 単に2つのWordPressがNAS上に別個に存在している構成なので、2つの独立したWordPressサイトと呼んでも間違いではありません。
ただし、「WordPressのマルチサイト構成」ではない ことには注意が必要です。マルチサイトはひとつのWordPressの中で複数のサイトを管理する仕組みで、今回のように個別にWordPressをインストールし、それぞれのドメインで運用している 場合は、それぞれが独立した「シングルサイト」です。
FAQ:
DBの修正を終えてWebブラウザで表示させたとき,表示はするが表示までの時間が長い場合,以下をチェックする.
DBの修正をミスしている.具体的には,site Aとsite Bのurlや/web/<folder A>と/web/<folder B>の置換のミス.以下のように設定を確認する.
テーブルのwp_optionsにあるsiteurl とhome を含むレコードが,サイトのurlであることを確認する.
テーブルのwp_postsのカラムのカラムのguidは,url+”?p=<数字>” (ex: harikir.diskstation.me/<wordpressがあるfoler名>/?p=1)となっているか.正しく置換できているか確認する.
どうしても正常にできない場合は,再度,DBをimportし直すところから始める.具体的には,当該DBを削除し,改めて当該DB名でDBを作成.その後,本編でも解説しているように,既存のDBのエクスポートファイルをimportして,修正作業をやり直す.
管理者でログインしてもログイン画面にならない場合の対処例
基本的な作業は,再度,DBをimportし直すところから始める.
DBのwp-optionsのsiteurlとhomeを修正する.
ブラウザから当該サイトを表示させてみる.フロントページの表示ができら以下に進む.
wp_postsのguidには,オリジナル(site 1)のurlが記載されているので,当該サイト(site 2)のurlに置換する.
ブラウザで表示していみる.
以上,ステップバイステップで確認していく.
環境変数(この記事中で説明しているwp-config.phpなど)をミスした場合,web server (portal)のレスポンスが失われることがある.その場合,MariaDB10の再起動,web stationのportalの再起動をしてやれば,レスポンスは回復する.NASのrebootは必要ない(はず).
MariaDB10のreboot : package centerからMariaDB10をクリック(openボタンではない)し,現れる「Open」ボタンの右横の矢印でStopを選択する.停止できたら,同じ要領でRunを選択する.
Portalのreboot: Web StationのWeb Portalをクリック.表示されるportalをクリックして選択.Actionボタンのリストからdisableをクリック.disableできたら,同じ要領でenableボタンをクリックする.
HTTPエラーコード一覧(代表例)
ステータスコード 名前 意味・原因の概要 要確認 404 Not Found 要求されたページが存在しない 。 portalがdisableに..htaccessが無い. 403 Forbidden アクセスが禁止されている 。 Access control profileなどで外部からのアクセス制限など. 500 Internal Server Error サーバー内部で予期せぬエラー が発生。 別のserverの.htaccessを使用している..htaccess内のpath指定が不整合
あとがき
複数サイトの稼働様子を見ながら,2つ目のサイトの構築を進めてきた.生じた問題については以下に示しているので参考にしてほしい.
2025/06/25, redis serverの確認(bridgeではなくhostになっていること)とwordpressのredis object cacheの設定(host設定とdatabase番号に関してwp-config.phpに設定する)がうまくいき,さらにapache 2.4がNASのreboot後に自動起動しないことについてtask schedulerで起動させることで数日の間,安定稼働している.
2025/06/20, 3台のweb server (multi domain)の安定稼働には,default serverのservice設定を適切にすることでレスポンスを失う事態にはまだいたっていない.このまま稼働を継続する.
2025/06/18, 結局,アクセスできなくなる現象(404)が生じた後に,MariaDB10をrebootすることで解消できることが分かった.その後,2~3時間すると,また,両サイトともに404エラーとなり,その後復旧しないというトラブルは引き続き変わらない.AI君と相談し原因・対策を検討した結果,2つ目のサイト用に別のuser2でのDB2を作成することで,当該トラブルは解消されている(12時間ほど2サイト構成で稼働).
2025/06/17, 昨日に続き,サイドInternetからいずれのHome Pageにもアクセスできなくなる現象が起きた.更に,Google Analiticsの新規ユーザー数が低下したことからアクセスされていないようだった.
2025/06/16, 1つ目(オリジナル)および2つ目もinternet(外部)からアクセスできなくなる現状が起きた.この日の対策は,Routerのrebootで解決したので,Routerが原因ではないかと考えてた.
編集履歴
2025/06/15, Mrはりきり



![Synology NAS : Multi Domain ~ 1つのNASの上に複数ドメイン名でサイトを構築する [2025/07/02追記]](https://harikiri.diskstation.me/wp-content/uploads/2020/12/36CF6F92-9A83-4A27-B301-25A96A970340.jpeg)


![Synology : RT6600ax ~ローカルから「当サイト」は閲覧できたが外からは見れなかった原因 (2025/06/12_9:00~10:40普通)になった対応の結果について [2025/06/16追記]](https://harikiri.diskstation.me/wp-content/uploads/2022/09/025BA3AC-1D91-4B46-9045-6D7DB8079731.jpeg)
![Excel : 3軸グラフ ~2軸の組み合わせで決まってくる3軸目の値から2つの軸の組み合わせの評価に使う~ [2025/06/11]](https://harikiri.diskstation.me/wp-content/uploads/2025/06/image-5.png)
![ギリシャヨーグルト 「バルティノ(はちみつ付)」にコーヒーを少々添加すると今までに無い新鮮な味 ~ 例えるとプリン風だが苦みが効いていて新感覚! [2025/06/11]](https://harikiri.diskstation.me/wp-content/uploads/2025/06/image-4.jpg)
![GxP: 新薬を開発したA社,自社および委託先が受けるpmdaによる適合性調査に備える・・・ [2025/06/09]](https://harikiri.diskstation.me/wp-content/uploads/2025/06/IMG_2610.png)

