タグ: flow-rAAV
-
[Bio-Equip] キャピラリー電気泳動システム – PA 800 Plus – SDS-PAGEおよびIEX分析
Post Views: 958 CESI 8000 Plus 抗体医薬を製造して品質を分析しますが、その分析項…
投稿者

-
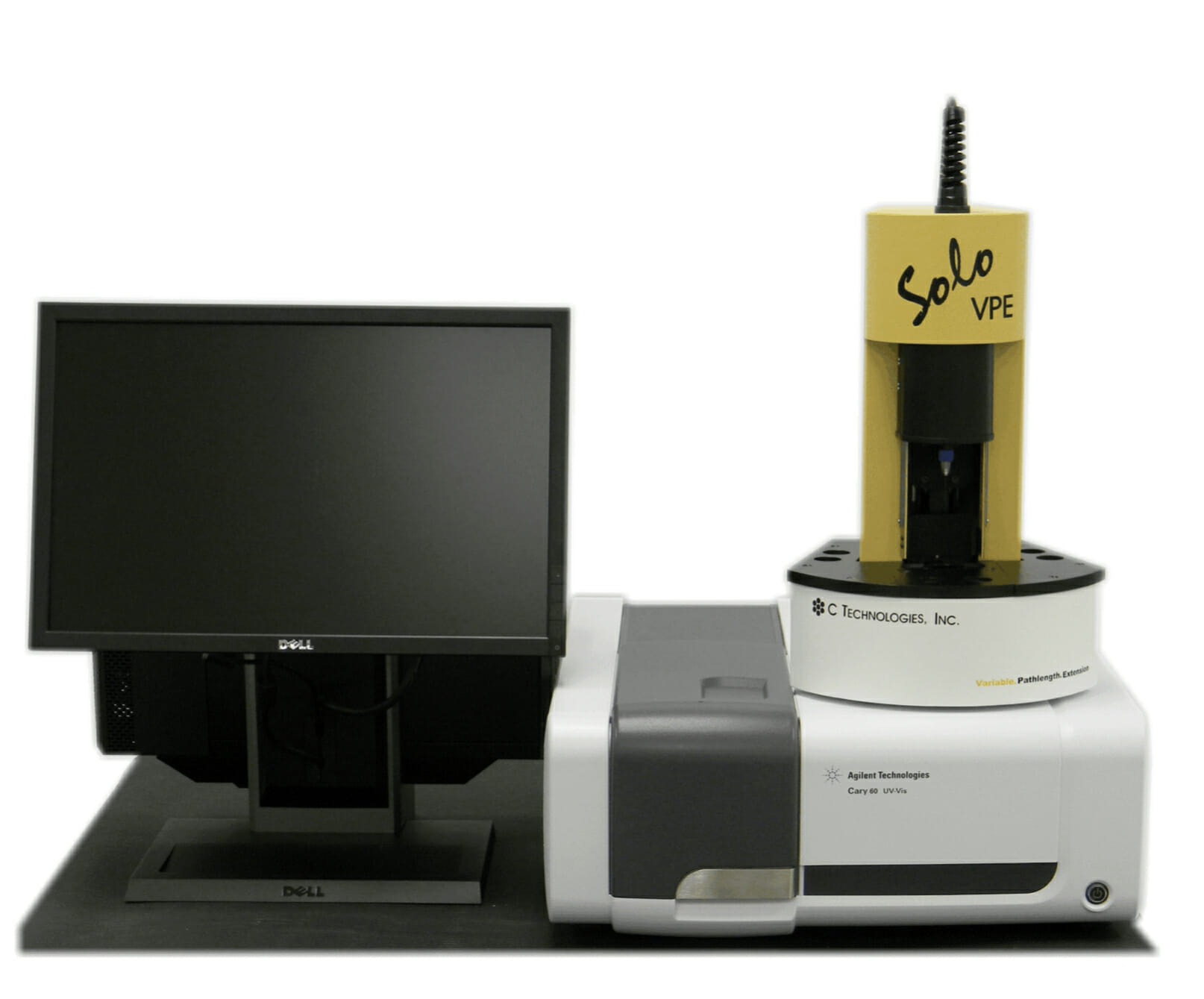
[Bio-Equip] SoloVPE – 高濃度タンパク質の無希釈測定 – GMP対応
Post Views: 1,004 SoloVPE 抗体医薬品のタンパク質濃度は、患者利便性から静注から皮下注…
投稿者

-
[Bio-Analysis] Comparability Test – バイオ医薬品における同等性/同質性 – ID8641 [2024/06/1]
Post Views: 722 はじめに Release Testとは、医薬品の製造が正常に実施され、それによ…
投稿者