![[WordPress] Encyclopedia プラグイン とAMP化](https://harikiri.diskstation.me/myblog/wp-content/uploads/2020/02/6BD71C35-5A28-4841-BE2E-C91EFA153F04.jpeg)
Encyclopedia プラグイン
機能
投稿で使用した用語に、吹き出しを付けるWordPressのプラグインです。
Encyclopediaプラグインは、ドイツ製(Deutsch)のTooltipプラグインです。吹き出しのことをTooltipというようです。単語を1つずつ定義します。ページの定義された単語がある場合、定義内容を吹き出しにできます。
Free版をしばらく使ってPro版にアップグレード(約5,500円)しました(2020/02/16)
AMPプラグインを導入していなければ、完全な機能を使うますが、AMPプラグインを導入している場合、AMPのルールに反するJavaScriptなどは削除リストに上がってきます。これを削除しないとAMPの速度向上の恩恵を受けられないので、削除するわけですが、Encyclopediaの機能の一部が使用できなくなります。
2022/01/02,
サイトのAMP化を更に進めるためるため、および、当該プラグインのPro版は1年間のサブスクリプションになっているので最新版へのアップデートが出来ないことで、Encyclopediaをアンインストールすることにしました。
登録してあるワードは、投稿(post)に変換していきます。その後全てを変換完了したらアンインストールする予定です。
吹き出しの機能は使えなくなりますが (iPadなどマウスカーソルがない場合はそもそも吹き出し機能は使えなかった)、その代わりの機能として、ページの末尾に記事の編集時にタグで指定したワードを利用して、そのワードを登録した記事(post)へのリンクをリストするようにphpでプログラムしました。
AMP対応ページで使用できなくなる機能
フォバー機能
iPhone/iPadでのクリックによるフォバーの代替機能が使えなくなる。これは、マウスカーソルが、登録単語の上に来た時(フォバー)、吹き出しを出す機能ですが、iPhone/iPadでは、マウスは通常使わないので、登録単語をタップした時に、吹き出しを出すようにする機能です。この機能が使えなくなります。ただし、タップした時に、フォバーは機能しないものの、登録の単語のページにはジャンプ可能です。Windowsなどマウスが使用可能なOSでは、フォバー機能は変わらず使えます。
設定
目標機能
- AMPプラグインを未導入の場合
- モバイルでは、マウスカーソルが無いため、マウスカーソルをリンクの上持ってくるという動作ができません。
- そこで、モバイルに対応させるために、クリックした場合でも、リンクにジャンプせず、吹き出しを表示する仕様にしたいと思います。
詳細設定
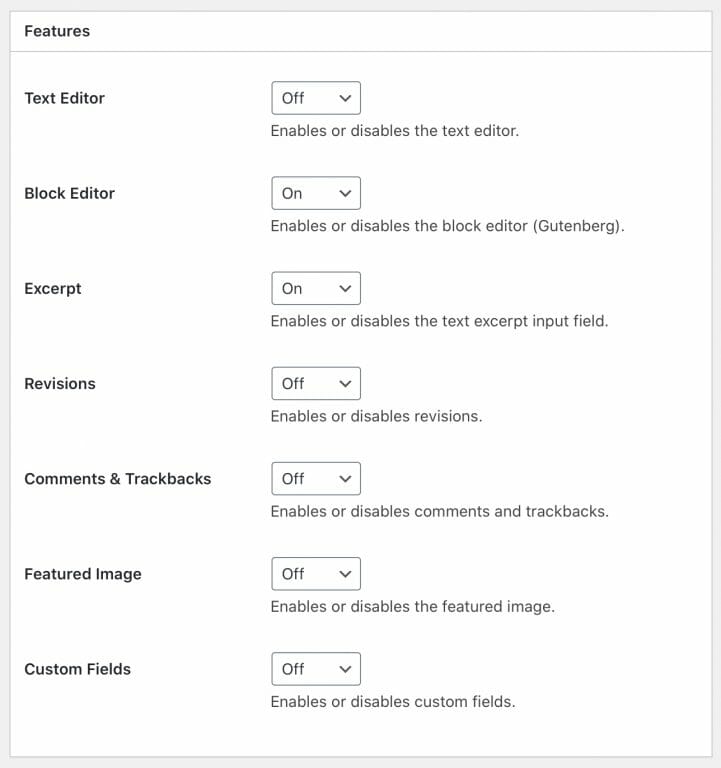
基本設定は、以下のように設定しています。
- ClassicのText Editorは基本的に使用しないので、Off
- Block Editorは、メインに使用しているので、On
- Excerpt (投稿の抜粋)は、以下の例では、Onになつていますが、複雑になるのでOffの方が良いかもしれません
- Revisionsは、Off
- Comments & Trackbacksは、Off
- Feature Imageは、Off
- Custom Fealdsは、何の項目か、良くわからないのでOff
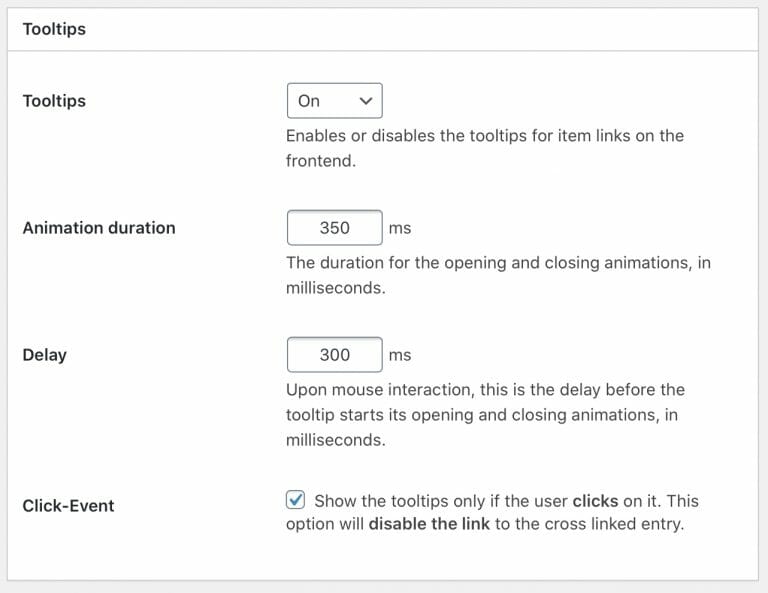
吹き出しの表示/リンクに飛ばない設定
- Tooltips : On
- Click-Event : ☑️
注意点
AMPプラグインを導入している場合、投稿がAMP有効になっていると目標機能は機能しません。
- クリックした時、リンクに飛んでしまいます。JavaScriptがAMPのルールに違反しているため、削除されているためと考えられます。
編集履歴
2020/03/07 はりきり(Mr) 2022/01/02,追記(AMP化を更に進めるためにアンインストールすることにした)
![[WordPress] Cool Timeline プラグイン[2020/02/25]](https://harikiri.diskstation.me/myblog/wp-content/uploads/2019/12/DFCE24D5-7AEA-4FF0-820C-D054B48E2D5C.jpeg)
![[WordPress] GT3 Photo & Video Gallery](https://harikiri.diskstation.me/myblog/wp-content/uploads/2020/01/4121A7BC-9ACF-465E-B248-9AD8849AC17B-1200x387.jpeg)
![[WordPress] Default feature image プラグイン [2020/02/25]](https://harikiri.diskstation.me/myblog/wp-content/uploads/2020/02/A5898744-76FF-4625-9F9D-8B59815212F9.jpeg)
![[WordPress] Easy Table of Contents プラグイン [2020/02/25]](https://harikiri.diskstation.me/myblog/wp-content/uploads/2020/02/714BA5C2-EC7E-467B-84FF-64EC744C632C.jpeg)
![[WordPress] UpdraftPlus プラグイン – バックアップと復元 [2021/03/07]](https://harikiri.diskstation.me/myblog/wp-content/uploads/2020/02/D8087769-5382-4A4B-A25D-9C5B03D1DF2A.jpeg)
![[WordPress] WP-Optimizer プラグイン [2020/09/24]](https://harikiri.diskstation.me/myblog/wp-content/uploads/2021/02/80F3D755-3019-49C6-BDAE-47D137C87826.jpeg)
![[WordPress] Lazy Load Optimize プラグイン](https://harikiri.diskstation.me/myblog/wp-content/uploads/2020/02/FAE70D6D-8157-4F99-AD24-5DCBA3E57BB6.jpeg)
![[WordPress] EWWW Image Optimize プラグイン](https://harikiri.diskstation.me/myblog/wp-content/uploads/2020/02/D4221C35-30D8-4173-8765-A97175E9E317.jpeg)
![[WordPress] Autoptimize プラグイン – サイト・速度改善 [2020/02/25]](https://harikiri.diskstation.me/myblog/wp-content/uploads/2020/02/autoptimize.jpg)